







 本文介绍了如何从NCBI的SequenceReadArchive(SRA)获取10xGenomics的单细胞测序数据,包括使用fastq-dump下载SRR文件,识别并转换文件命名,以满足CellRanger的处理要求。
本文介绍了如何从NCBI的SequenceReadArchive(SRA)获取10xGenomics的单细胞测序数据,包括使用fastq-dump下载SRR文件,识别并转换文件命名,以满足CellRanger的处理要求。
1. 获取数据的SRR*
根据参考文献提供的GEO数据链接,直接在浏览器打开,或者NCBI官网检索GSE号,在数据网址的最下方点击SRA Run Selector链接跳转至新的页面,该页面包含原始fastq文件的SRR编号,和样本的一些基本信息。
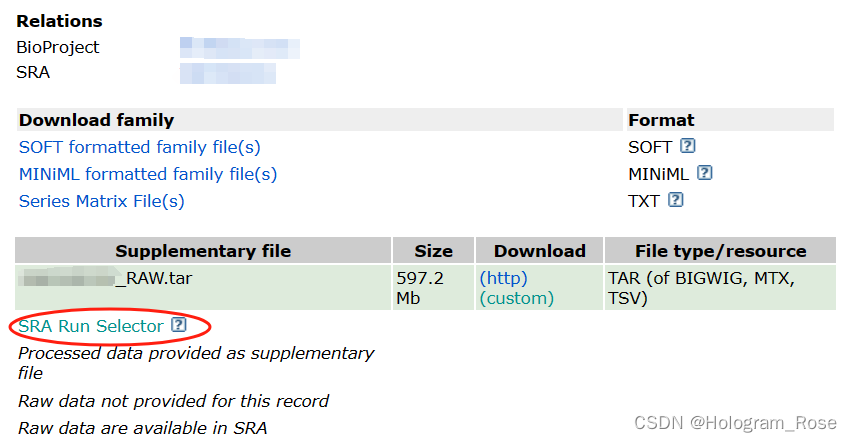
2. 使用fastq-dump下载数据
以SRR9264343为例
fastq-dump --split-files --gzip SRR9264343
下载完成后有以下三个文件,Cell Ranger mkfastq处理之后,一般会产生3个fastq.gz文件,分别是I1文件,8bp的样本barcodes;R1文件16bp feature barcodes + 10 bp/12 bp UMI; R2文件转录组的reads文件。
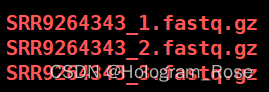
从SRA Run Selector页面,单击单个样本的SRR*,可以跳转至以下页面并点击Data access,根据Original format处的信息,可以清晰地分辨下载后3个的文件分别对应的具体文件。
 最低0.47元/天 解锁文章
最低0.47元/天 解锁文章


















 1698
1698

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









