










在药物研发领域,化学空间的广阔性意味着无数潜在化合物尚待探索和发现。然而,这些化合物中只有极少数具备治疗疾病的潜力。面对如此庞大的化合物库,如何快速、高效地筛选出具有潜在应用价值的药物分子,始终是该领域的一大挑战。其中,高通量筛选和虚拟筛选是常用的两种策略,它们各自具有独特的优势和工作机制,共同推动着药物研发进程的加速。
高通量筛选 VS 虚拟筛选
高通量筛选 (High-Throughput Screening, HTS): 也叫实验筛选,是使用生物物理、生物化学等方法鉴别小分子和蛋白质的相互作用,从而筛选出对蛋白质功能有调节作用的先导化合物。
虚拟筛选 (Virtual Screening, VS): 指在进行生物活性实验筛选之前,根据预先设定的条件,在计算机上对化合物库中的分子进行预筛选,以识别出最有可能与靶标结合的小分子。这一过程大大减少了实际筛选的化合物数目,同时提高先导化合物的发现效率。
表 1. 高通量筛选与虚拟筛选对比
| 特点 | 高通量筛选 (HTS) | 虚拟筛选 (VS) |
|---|---|---|
| 基础 | 体外活性测试 | 计算机模拟分析 |
| 时间与成本 | 成本高,需要大量实验资源 | 成本较低,初期筛选快速 |
| 数据需求 | 化合物库、高效实验平台、生物标志物 | 靶点结构、配体结构、对接打分函数等 |
| 规模 | 数万到十万级化合物实验筛选 | 数百万级化合物虚拟筛选 |
| 命中率 | 0.01-0.001% | 2-24% |
| 精度 | 生物实验直接测试,结果准确性高但可能有误差 | 依赖于建模质量和算法,可能产生虚假阳性或遗漏活性化合物 |
-
虚拟筛选 适合作为 早期筛选工具,筛选潜在活性分子后结合高通量筛选验证,降低实验成本并提高效率。
-
高通量筛选 更适用于 后期验证 及直接探索复杂生物系统的活性分子库。
高通量筛选与虚拟筛选两种方法的结合在现代药物开发中尤为常见,利用虚拟筛选筛选初步候选化合物,再用高通量筛选对候选分子进行验证和优化,从而大大提高新药发现的效率和成功率。
虚拟筛选常用方法
常用的虚拟筛选方法可以分为两大类:基于 配体的虚拟筛选 和基于 受体/结构的虚拟筛选。
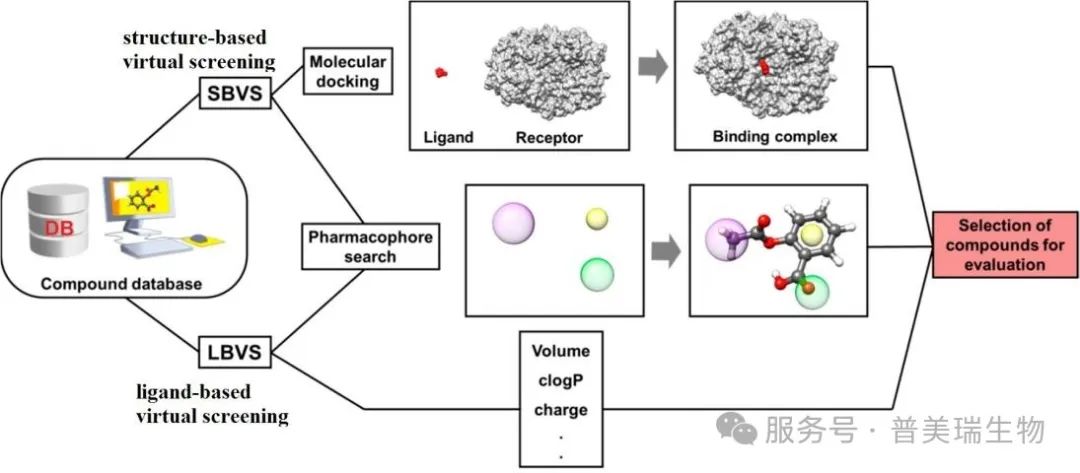
图 1. 虚拟筛选基本流程和方法 [1]
基于配体的虚拟筛选
基于配体的虚拟筛选 (Ligand based virtual screening, LBVS) 依赖于 已知配体的化学和结构特性,通常认为 具有相似结构的化合物往往具有类似的药理活性。在靶标未知或蛋白结构尚未解析的情况下,基于配体的虚拟筛选是一种有效的药物发现策略。该方法利用已知的活性配体作为模板或探针,从庞大的化合物库中筛选出与其 具有相似结合特征 或 作用机制 的潜在药物候选分子。
基于配体的虚拟筛选方法包括:
-
基于结构相似度的虚拟筛选: 假设具有相似化学结构的分子可能表现出相似的生物活性。
当有高活性先导化合物但缺乏靶点结构时,计算该化合物的分子指纹(如 MACCS 或 ECFP),通过 Tanimoto 系数衡量该化合物与其他化合物之间的相似性,从而找到具有类似特性的新的化合物分子。
-
基于定量构效关系 (QSAR) 的虚拟筛选: 基于已知分子的结构和生物活性数据,利用化学信息学工具,将化合物的结构信息转化为可量化的描述符(如分子量、原子组成、化学键类型、分子形状、电荷分布等),作为建立模型的输入变量。
采用多元线性回归 (MLR)、偏最小二乘 (PLS) 等方法,基于提取的特征和已知的生物活性数据,训练并建立 QSAR 模型。
将构建好的 QSAR 模型应用于大规模的化合物库,预测每个化合物的生物活性。
根据预设的活性阈值,筛选出具有高预测活性的候选分子,作为后续实验验证的优先级对象。
-
基于药效团模型的虚拟筛选: 通过提取活性化合物共有的药效团特征(如疏水中心、氢键供体/受体、芳香中心等),构建药效团模型,并以此为模板在大规模化合物库中进行匹配搜索,以识别出具有相似药效团特征的潜在活性分子。
基于受体/结构的虚拟筛选
近年来,随着 结构解析技术 (如 X 射线晶体学、核磁共振成像 NMR、冷冻电镜 Cryo-EM) 和 AlphaFold 的迅速发展,越来越多的蛋白质三维结构被解析,这极大地推动了基于结构的虚拟筛选技术的发展。基于结构的虚拟筛选 (Structure based virtual screening, SBVS) 通过靶蛋白的三维结构来预测配体的结合模式,从而找到潜在的药物分子。主要通过 分子对接技术 将候选分子与靶蛋白的活性位点进行对接,评估分子间的相互作用能、结合模式及可能引发的构象变化,从而筛选出与靶蛋白具有高亲和力和特异性的候选药物分子。
基于受体/结构的虚拟筛选方法包括:
-
基于分子对接、分子动力学模拟、结合自由能计算的虚拟筛选: 首先利用分子对接技术,通过计算模拟将候选分子与靶标的活性位点进行初步匹配,筛选出空间构型上合适的分子。
随后,采用分子动力学模拟,动态地评估这些分子在靶标结合口袋中的稳定性及相互作用模式,进一步筛选出动态行为合理的分子。
结合自由能的计算则提供了分子与靶标结合强度的量化指标,有助于精确筛选出具有高亲和力的分子。
-
基于受体药效团模型的虚拟筛选: 在未知配体结构的情况下,通过对受体活性位点特征结构的深入理解,构建出能够代表受体关键相互作用特性的药效团模型。
利用该模型对化合物库进行大规模筛选,从而可以高效地识别出那些能够与药效团模型良好匹配的候选分子,这些分子很可能具有与受体结合并触发预期生物学效应的能力。
-
基于片段设计的虚拟筛选: 首先构建一个包含大量小分子片段的数据库,这些片段通常具有特定的官能团或化学特性,能够与靶标的特定区域产生相互作用。
随后,通过分子对接技术筛选出那些能够与靶标形成稳定且特异性结合的片段。
基于筛选出来的片段,采用片段设计的方法(比如片段生长、片段连接等),构建出具有更高亲和力和生物活性的完整分子。
小结
虚拟筛选作为一种高效的药物发现工具,凭借其强大的计算能力,能够精准地预测化合物的生物活性,从而显著地加速了新药从初步发现到深入开发的整个进程。这一技术的运用,不仅大大地缩减了药物研发所需的时间成本,还提升了研发过程的整体效率。随着近年来计算能力的持续飞跃、生物信息学领域的蓬勃发展以及实验技术的不断革新,虚拟筛选技术在药物设计这一关键领域内展现出了愈发重要的影响力,并持续推动着该领域的边界拓展。
普美瑞生物科技计算平台提供了包括基于配体和受体在内的多个在线虚拟筛选工具。其中,基于配体的虚拟筛选工具包括3D相似性搜索(根据用户上传的分子结构,在百万级别的小分子化合物库进行高效的3D结构相似性匹配与检索,快速筛选出与上传分子三维结构高度类似的潜在活性分子)和基于配体的靶点搜寻(根据用户上传的目标分子结构,在已知靶点的活性化合物库中进行高效的相似性匹配与检索,从而获得潜在作用靶标相关信息)。基于受体的虚拟筛选工具包括基于分子对接的虚拟筛选(基于深度学习算法的蛋白-小分子对接工具用于化合物库筛选,从而获得潜在活性小分子)和蛋白相互作用预测(通过搜索同源蛋白互作对象的方式,预测目标蛋白潜在的互作对象蛋白)。如想详细了解这些工具的介绍和使用,可登录普美瑞生物科技计算平台(www.pumeirui.com),查看相关工具的使用说明并申请试用。
参考文献
[1] 瀚鸿化学. 基于结构的药物分子虚拟筛选以及药物与靶点作用机制的研究. 2023.
欢迎联系我们
📞Tel: 0519-68007026
📧E-mail: info@pumeirui.com
🌐欢迎登录www.pumeirui.com

















 225
225

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









