







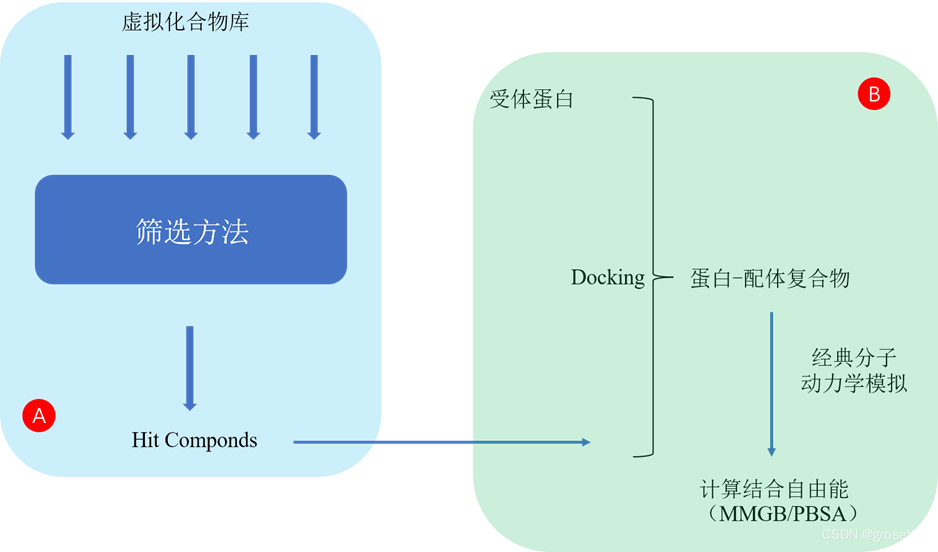
通常所说的虚拟筛选是图中A区域所示的过程。在获得Hit Componds后,一般有条件的可以直接用实验验证。但为了提升阳性率,降低实验成本,也可以继续进行B区域所示过程,运用分子动力学模拟的手段进一步过滤Hit Componds(但此步往往耗时较长,操作也更为繁琐,门槛更高)。
- 虚拟化合物库来源途径很多,免费的数据库有Zinc、PubChem、ChEMBL、DrugBank、ChemDB等,商业数据库有陶术、百灵威等。下载的虚拟化合物库文件格式往往是sdf或mol2后缀,切不可选用pdb格式(pdb格式专为蛋白质准备,对于小分子而言会忽略掉很多成键信息)。
- 筛选方法分为以下几类:①基于分子对接的虚拟筛选(最常用);②基于药效团的虚拟筛选(快速);③基于分子相似性的虚拟筛选;④基于定量构效关系模型QSAR筛选(精度取决于模型精度)。
- 基于分子对接的虚拟筛选:即批量分子对接。一般步骤有:①小分子库预处理(这一步主要是为了去除盐离子、生成同分异构/立体异构体、质子化等);②受体蛋白分子预处理(此步为了去除水分子、辅助结晶分子、结构松弛、质子化等);③在受体分子上指定配体结合位点;④筛选。
(需要注意的是,虚拟筛选打分数值直接用于比较的价值不大,其主要起到分子富集的作用,这是因为现有的打分函数大多不精确导致的。筛选到的一众化合物还需要根据经验判断挑选出合适的分子进行实验验证,挑选的依据有:观察配体分子的结合姿势是否更“自然”、形成的相互作用是否丰富等。但如果在虚拟筛选之后采用了分子动力学模拟计算蛋白-配体结合自由能,这样的计算结果才更具有横向比较意义,也能进一步过滤分子。) - 基于药效团的虚拟筛选(快速):一般步骤:①小分子库预处理;②提取药效团模型(这个需要根据经验判断,选择保留/去除哪些药效团);③筛选。
- 基于分子相似性的虚拟筛选:这种方法使用的前提需要有确定活性的小分子。将此小分子视为模板,从化合物库中过滤出与之相似的分子(通过提取分子指纹来比较相似性,提取分子指纹的算法有许多,包括Topological Fingerprints、MACCS、Atom Pairs and Topological Torsion、Morgan Fingerprints)。
- 基于定量构效关系模型(QSAR)筛选:首先通过文献搜集已报道有活性的分子,记录下这些分子的结构及活性数值(需要同一类活性数据,如同为IC50或同为抑制率)。从搜集到的分子结构中计算或预测它们的物理化学性质,如logP、TSA、分子量等,将这些计算出的信息和活性值一同作为模型的“训练数据”。一般来说,理想的QSAR模型可直接用于预测其他任何化合物的活性。


















 2313
2313

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











