






1. 从ncbi下载测序数据
SRA链接:https://www.ncbi.nlm.nih.gov/sra
检索所需的项目,这里以Whole genome sequencing of ExPECs (SRR24129389)为例。
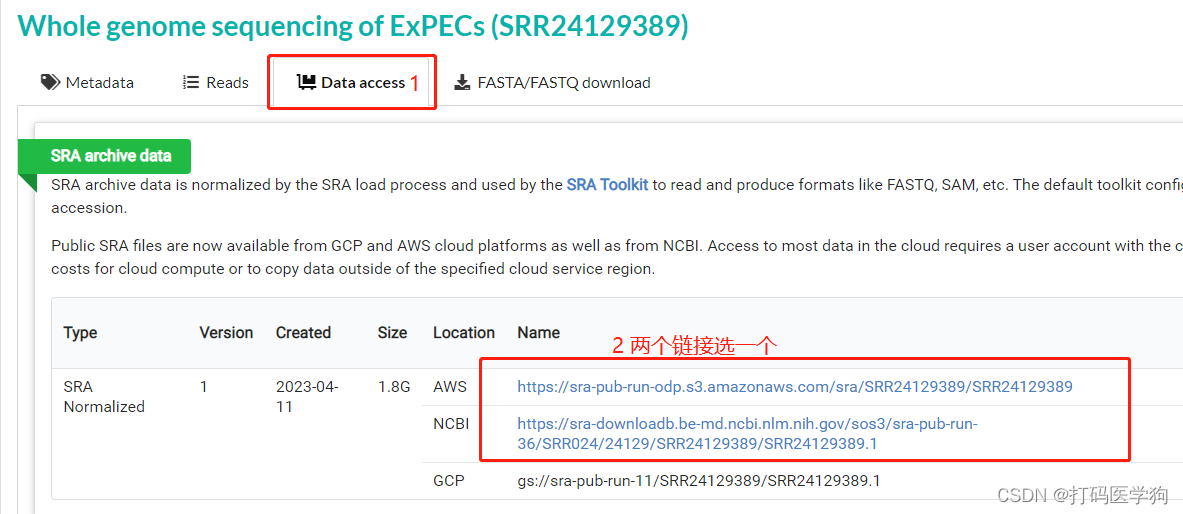
wget -c -t 0 -O ./SRR24129389.sra https://sra-pub-run-odp.s3.amazonaws.com/sra/SRR24129389/SRR24129389
# -c -t 配合使用可以防止下载数据的过程中链接中断的问题
# -O则可以指定下载路径和文件名。
2. 安装sratoolkit
方法1
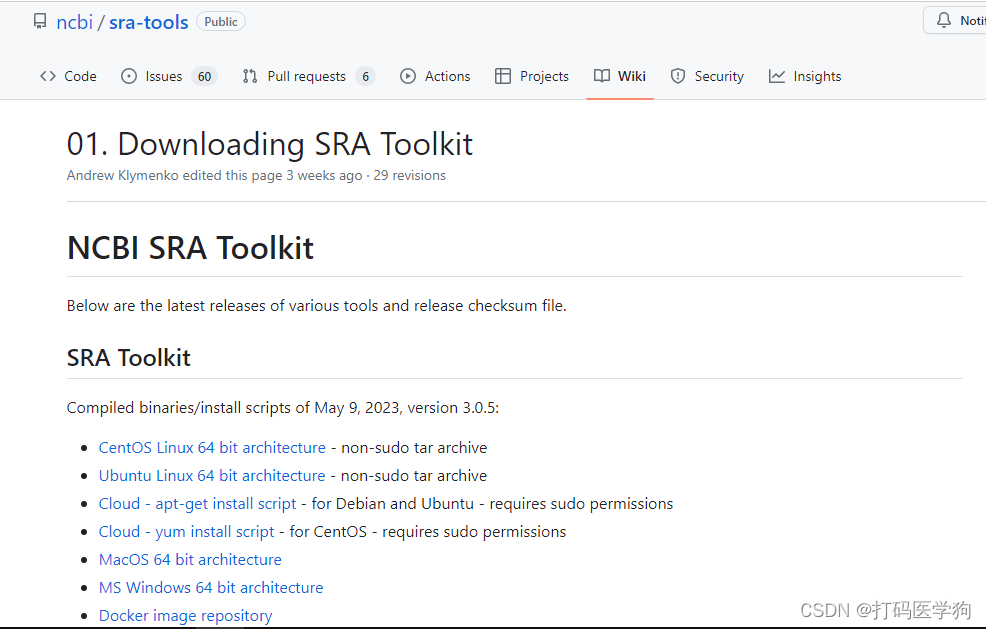
NCBI中各个操作系统下载链接:https://github.com/ncbi/sra-tools/wiki/01.-Downloading-SRA-Toolkit
##1.下载
wget --output-document sratoolkit.tar.gz https://ftp-trace.ncbi.nlm.nih.gov/sra/sdk/3.0.5/sratoolkit.3.0.5-ubuntu64.tar.gz
##2.解压
tar -vxzf sratoolkit.3.0.5-ubuntu64.tar.gz
##3.配置环境
echo "export PATH=/home/shpcv2_kvce3/software/sratoolkit.3.0.5-ubuntu64/bin:\$PATH ">>~/.bashrc
source ~/.bashrc
##4.验证
which fastq-dump
#输出 /home/shpcv2_kvce3/software/ratoolkit.3.0.5-ubuntu64/bin/fastq-dump
##5.测试
fastq-dump --stdout -X 2 SRR390728
#输出以下:
#Read 2 spots for SRR390728
#Written 2 spots for SRR390728
#@SRR390728.1 1 length=72
#CATTCTTCACGTAGTTCTCGAGCCTTGGTTTTCAGCGATGGAGAATGACTTTGACAAGCTGAGAGAAGNTNC
#+SRR390728.1 1 length=72
#;;;;;;;;;;;;;;;;;;;;;;;;;;;9;;665142;;;;;;;;;;;;;;;;;;;;;;;;;;;;;96&&&&(
#@SRR390728.2 2 length=72
#AAGTAGGTCTCGTCTGTGTTTTCTACGAGCTTGTGTTCCAGCTGACCCACTCCCTGGGTGGGGGGACTGGGT
#+SRR390728.2 2 length=72
#;;;;;;;;;;;;;;;;;4;;;;3;393.1+4&&5&&;;;;;;;;;;;;;;;;;;;;;<9;<;;;;;464262
参考官方指南:
https://github.com/ncbi/sra-tools/wiki/02.-Installing-SRA-Toolkit
方法2
使用conda安装, 首先先唤醒conda
source {你的conda安装目录/bin/activate}
# 我的代码 source ~/miniconda/bin/activate
conda install -y sra-tools
或
sudo apt install sra-toolkit
在收集了SRA编号后,还可以直接获取下载链接
srapath SRR24129389
## 输出为 https://sra-pub-run-odp.s3.amazonaws.com/sra/SRR24129389/SRR24129389
3. 将sra格式数据转化为fastq格式数据
方法一:使用fastq-dump
fastq-dump --gzip --split-3 -O ${outdirectory} SRR24129389.sra
##参数
#--gzip :输出gz格式压缩文件,节省空间,稍微多费点时间
#-O ${directory} :设置输出的文件间路径,outdirectory改为相应路径
#--split-3 :不知道sra是单端还是双端,默认使用--split-3
##例 fastq-dump --gzip --split-3 -O trans SRR24129389.sra
##trans是我设置的输出文件夹
方法二:使用fasterq-dump
使用差不多,参数上多了线程数 -e
fasterq-dump -p -e 24 --split-3 -O ${outdirectory} SRR24129389.sra
#-p 可以显示进程
#-e 24 使用24个线程
## fasterq-dump -p -e 4 --split-3 -O trans SRR24129389.sra
##trans是我设置的输出文件夹
总结一下,fasterq-dump速度可以完胜fastq-dump,值得注意的是,fasterq-dump没有压缩选项,而fastq-dump可以直接输出gz压缩fq文件
4. FastQC 安装
4.1 FastQC简介
FastQC是一款基于Java语言设计的软件,目前可以直接下载免费使用,一般在Linux环境下使用命令行执行程序,它可以快速地多线程地对测序数据进行质量控制(Quality Control),还能进行质量可视化来查看质控效果。运行一段时间以后,会出现报告。使用浏览器打开后缀是html的文件,这就是图表化的fastqc报告。
4.2 首先安装Java环境
#查看是否已安装了Java
which java
java -version
#安装Java
sudo apt install default-jre
4.3 基于conda安装FastQC
source /miniconda/bin/activate #启动唤醒conda
conda create -n fastqc #首先创建fastqc环境,输入y
conda active fastqc #进入fastqc环境
conda install -c bioconda fastqc #安装fastqc
fastqc --version ##查看是否安装成功
fastqc --help ##查看参数
4.4 使用fastqc进行质量检测
#单个文件处理
fastqc 样本名称
#批量文件处理
fastqc 样本1 样本2 … -o 文件夹 #默认输出文件夹为输入文件所在的位置
#或
fastqc *.fastq -o 文件夹 #默认输出文件夹为输入文件所在的位置
生成.html网页文件和.zip文件

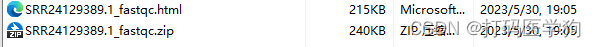
















 1974
1974











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









