









1,数据准备
类似于在R包seurat使用中介绍的单样本分析,
scanpy中的这一部分也是使用经典的PBMCs数据集进行演示,
事实上scanpy的作者也表示这部分内容实质上是seurat流程的python重现。
PBMC3K是一个10X Genomics提供的测试数据,可以到10X的官网上去下载
https://www.10xgenomics.com/what-is-single-cell-rna-seq
分析方面也有教程相关的网页
https://www.10xgenomics.com/analysis-guides
下载链接在于:
http://cf.10xgenomics.com/samples/cell-exp/1.1.0/pbmc3k/pbmc3k_filtered_gene_bc_matrices.tar.gz
如果在shell中,或者是在jupyter notebook的magic command中,可以使用终端命令如下
!mkdir data
!wget http://cf.10xgenomics.com/samples/cell-exp/1.1.0/pbmc3k/pbmc3k_filtered_gene_bc_matrices.tar.gz -O data/pbmc3k_filtered_gene_bc_matrices.tar.gz
!cd data; tar -xzvf ./pbmc3k_filtered_gene_bc_matrices.tar.gz
!mkdir write
用wget或者是用curl
解压之后的数据见下:
主要是其中的这3个文件:
关于barcodes.tsv、genes.tsv、matrix.mtx文件的详细介绍,
类似于CellRanger的输出文件:

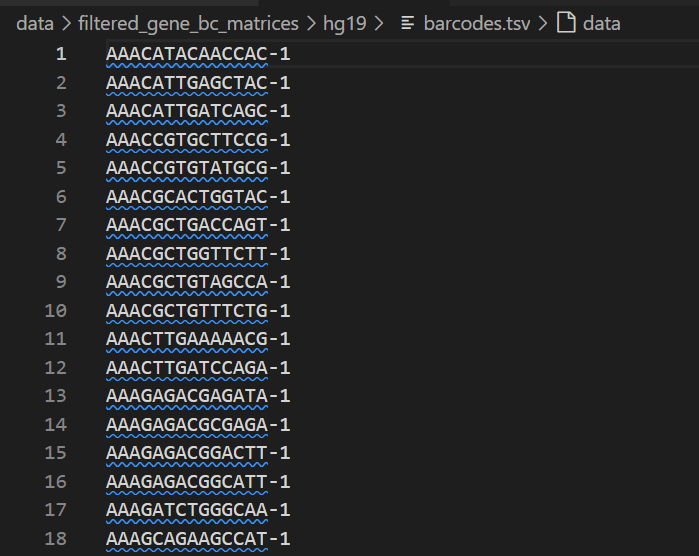
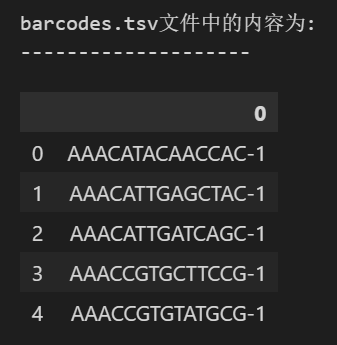
(1)barcodes.tsv:每行是一个细胞,1个样本

# 使用 base R 读取
barcodes <- read.table('/home/liur/hg19/barcodes.tsv', header=FALSE, sep='\t')
# 使用 tidyverse 读取
library(readr)
barcodes <- read_tsv('/home/liur/hg19/barcodes.tsv', col_names=FALSE)
# 查看前几行数据
head(barcodes)

比如说上面这个示例数据就有2700个cell,也就是2700个样本
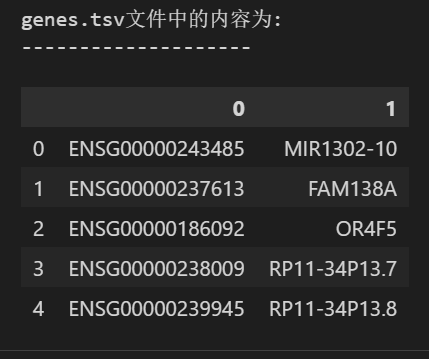
(2)gens.tsv
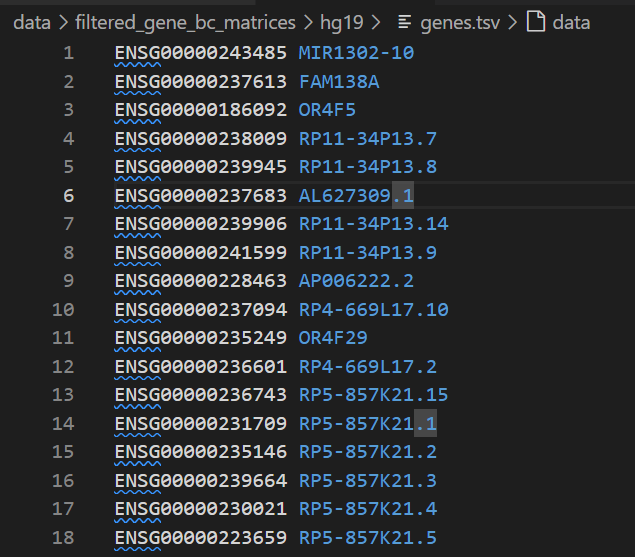


可以看到第1列就是ensembl的gene id,第2列是常用的symbol,然后一共是32738个gene,一般就是samplexfeature,所以可以说是feature文件
# 使用 base R 读取
genes <- read.table('/home/liur/hg19/genes.tsv', header=FALSE, sep='\t')
# 使用 tidyverse 读取
library(readr)
genes <- read_tsv('/home/liur/hg19/genes.tsv', col_names=FALSE)
# 查看前几行数据
head(genes)
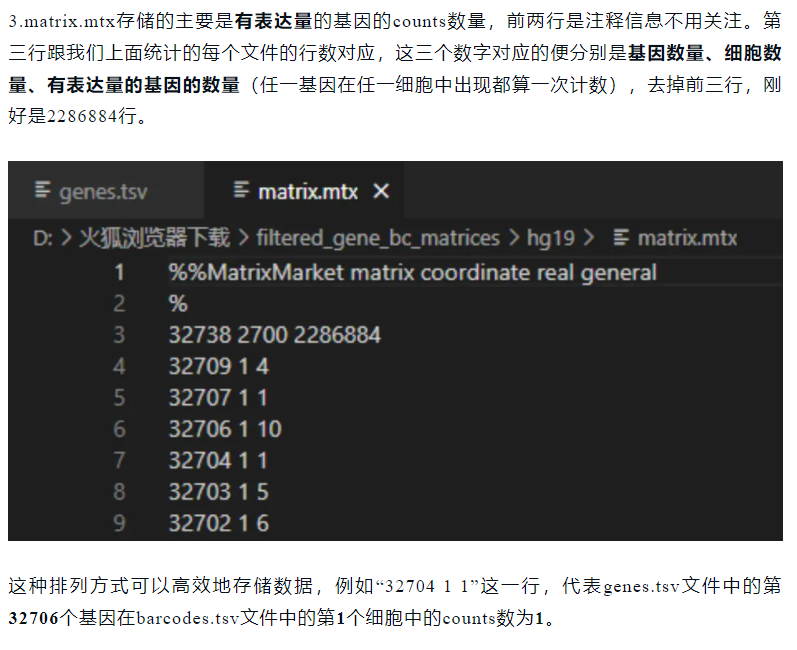
(3)matrix.mtx
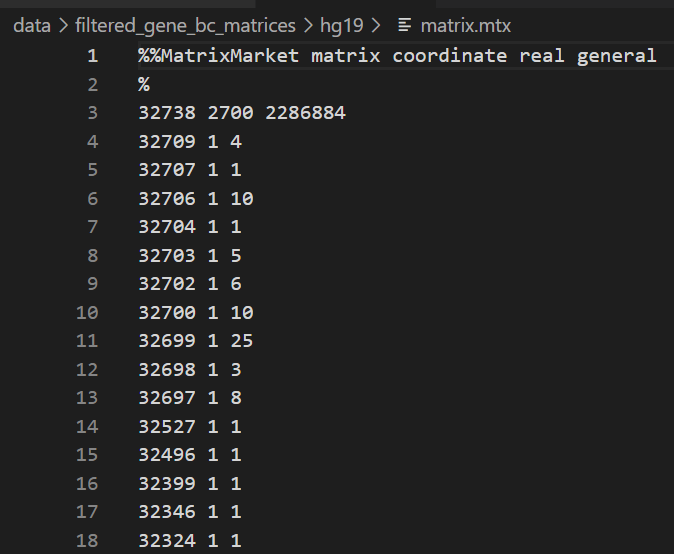

总之这里存储的就是稀疏矩阵的原始值(cellranger下机),
然后这个矩阵,按照scanpy以及python的习惯是sample x feature,也就是annData中的cell x gene,
当然在seurat中是转置就是gene x cell
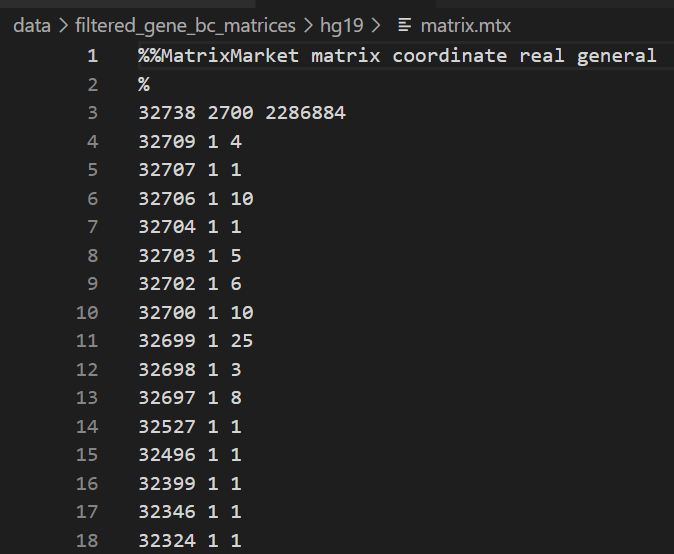
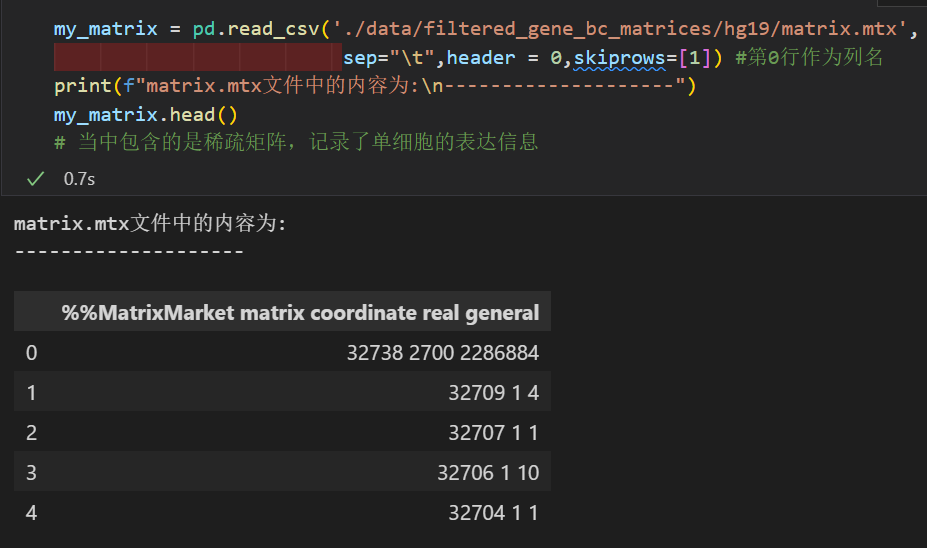
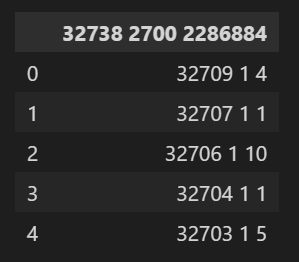
所以我们这里的值,大概意思就是这个矩阵有32738行,2700列,然后一共有2286884个非零值(也就是稀疏矩阵中非零值),
然后前面读取的gene数目以及cell数目tsv文件我们可以得知,
32738是gene,2700是cell,
所以这个矩阵是gene x cell x value的矩阵,
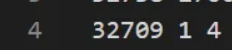
举例来说,也就是gene文件中的第32709行,cell文件中的第1列,对应的值是4


library(Matrix)
# 读取 matrix.mtx 文件
matrix <- readMM("/home/liur/hg19/matrix.mtx")
# 查看矩阵的维度
dim(matrix)
# 查看矩阵的内容
matrix[1:5, 1:5]
2,数据文件结构
my_barcode = pd.read_csv('./data/filtered_gene_bc_matrices/hg19/barcodes.tsv',
sep="\t",header = None) #没有列名
print(f"barcodes.tsv文件中的内容为:\n--------------------")
my_barcode.head() ## 顾名思义,这个文件中包含的是细胞的barcode信息
因为没有R中的read_tsv,所以统一使用read_csv
my_genes = pd.read_csv('./data/filtered_gene_bc_matrices/hg19/genes.tsv',
sep="\t",header = None) #同样是没有列名的
print(f"genes.tsv文件中的内容为:\n--------------------")
my_genes.head()
# 共包含两列,一列为基因ID,一列为基因SYMBOL
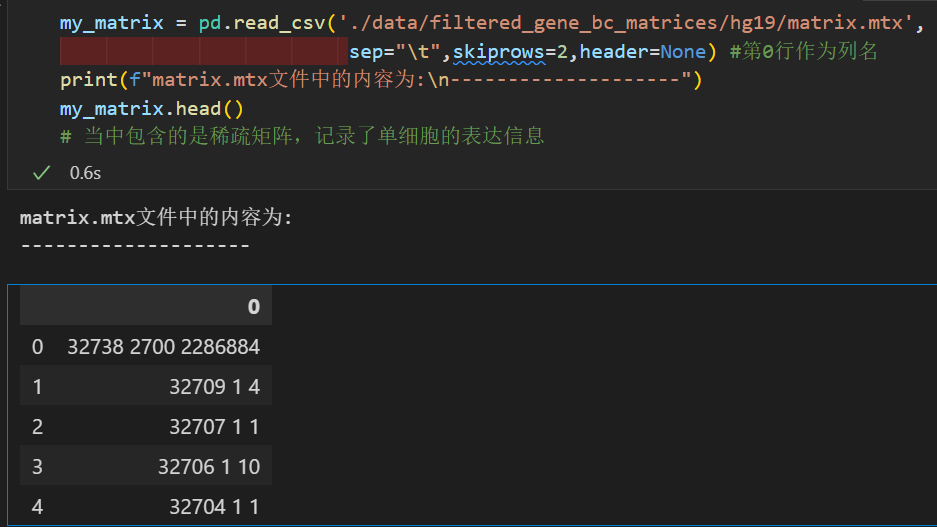
my_matrix = pd.read_csv('./data/filtered_gene_bc_matrices/hg19/matrix.mtx',
sep="\t",header = 0) #第0行作为列名
print(f"matrix.mtx文件中的内容为:\n--------------------")
my_matrix.head()
# 当中包含的是稀疏矩阵,记录了单细胞的表达信息

如果只是想跳过第1行(0-index下的line1,也就是上面原始数据中的%符号)
my_matrix = pd.read_csv('./data/filtered_gene_bc_matrices/hg19/matrix.mtx',
sep="\t",header = 0,skiprows=[1]) #第0行作为列名
print(f"matrix.mtx文件中的内容为:\n--------------------")
my_matrix.head()
# 当中包含的是稀疏矩阵,记录了单细胞的表达信息
或者:
使用comment参数跳过
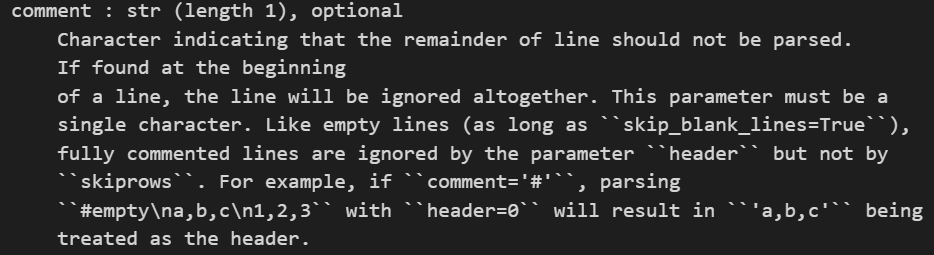
import pandas as pd
# 读取 Matrix Market 格式的文件,将第一行作为列名,并跳过以 % 开头的注释行
my_matrix = pd.read_csv('./data/filtered_gene_bc_matrices/hg19/matrix.mtx',
sep="\t", header=0, comment='%')
# 打印读取的数据
my_matrix.head()
主要是关注一些优先级之类的:
然后注意是0-index的line number

事实证明这个不是tsv文件
3,数据获取
(1)导包
import numpy as np
import pandas as pd
import scanpy as sc
(2)全局设置
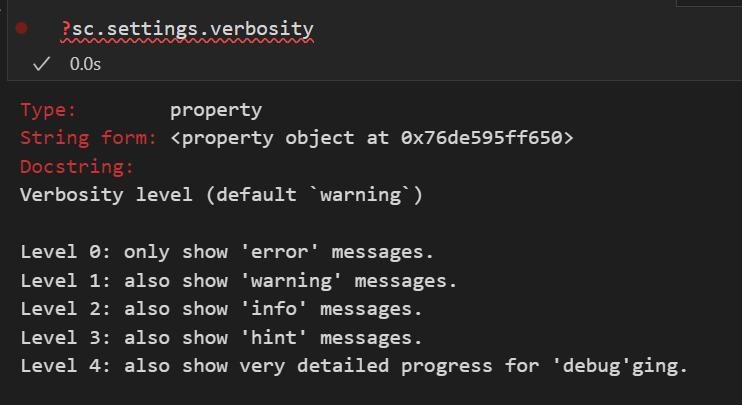
# 进度条设置
sc.settings.verbosity = 3
# verbosity: errors (0), warnings (1), info (2), hints (3)
# 输出日志只展示开头信息,用于打印可能影响数值结果的各种依赖包的版本信息
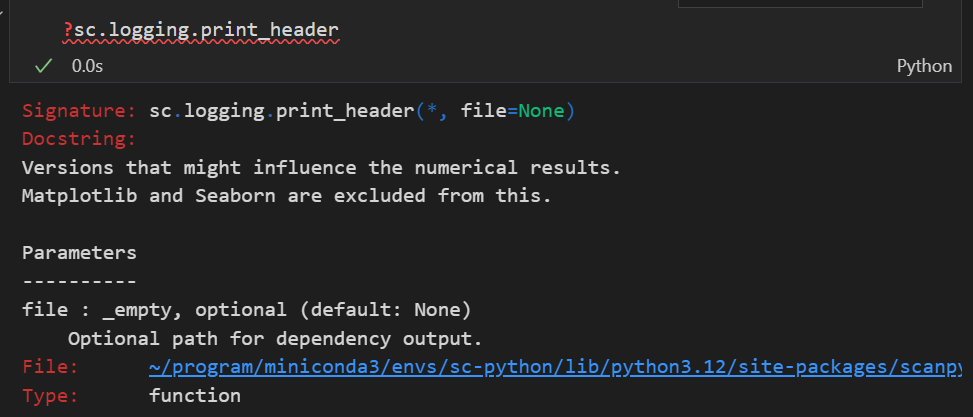
sc.logging.print_header()
# 输出图片分辨率设置,这里演示为80,建议实际运行时设置为300(一般投稿要求)
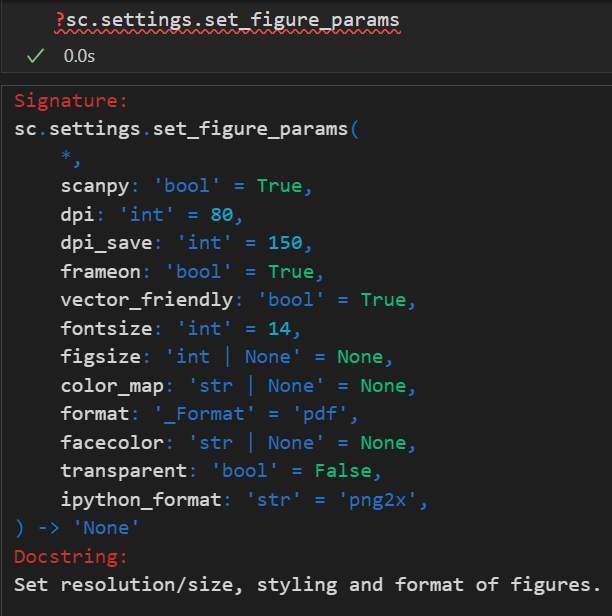
sc.settings.set_figure_params(facecolor='white', dpi=100, fontsize=10, dpi_save=300, figsize=(10, 7), format="pdf")
# 这个函数是设置图片的尺寸,这个跟R语言中就很不一样,一般R语言会在输出环节再设置。
# scanpy: 如果为True,使用Scanpy的默认matplotlib设置。
# dpi: 图像显示的分辨率。
# dpi_save: 保存图像的分辨率。
# frameon: 是否在图像周围显示框架。
# vector_friendly: 是否生成对矢量图友好的输出。
# fontsize: 图形中使用的字体大小。暂时设置为weightxheight!!!!!!
# figsize: 图形的尺寸,如果为None,则使用默认尺寸。
# color_map: 使用的颜色映射。后续美化的时候可以设置!!!!!!!!
# format: 保存图形时使用的格式。
# facecolor: 图形的背景颜色,如果为None,使用默认颜色。
# transparent: 是否使图形背景透明。
# ipython_format: 在Jupyter Notebook中显示图形时使用的格式。
# 设置输出文件名:
results_file = 'write/pbmc3k.h5ad'
设置 scanpy 的日志输出详细程度:暂时设置为3,最详细的设置为4
打印 scanpy 的日志头信息,包括版本号和依赖库的信息,有助于记录分析环境的详细信息,便于调试和复现
设置 scanpy 输出图片的参数

这里报了一个小warning,还是有必要处理一下:
TqdmWarning: IProgress not found. Please update jupyter and ipywidgets. See https://ipywidgets.readthedocs.io/en/stable/user_install.htm
输出结果为
scanpy==1.10.4 anndata==0.11.3 umap==0.5.7 numpy==2.1.3 scipy==1.15.1 pandas==2.2.3 scikit-learn==1.6.1 statsmodels==0.14.4 igraph==0.11.8 pynndescent==0.5.13
(3)数据读取
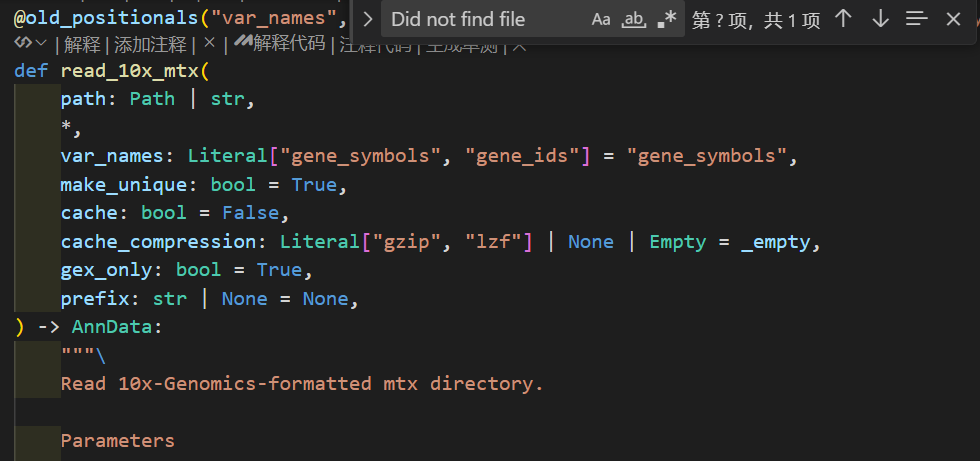
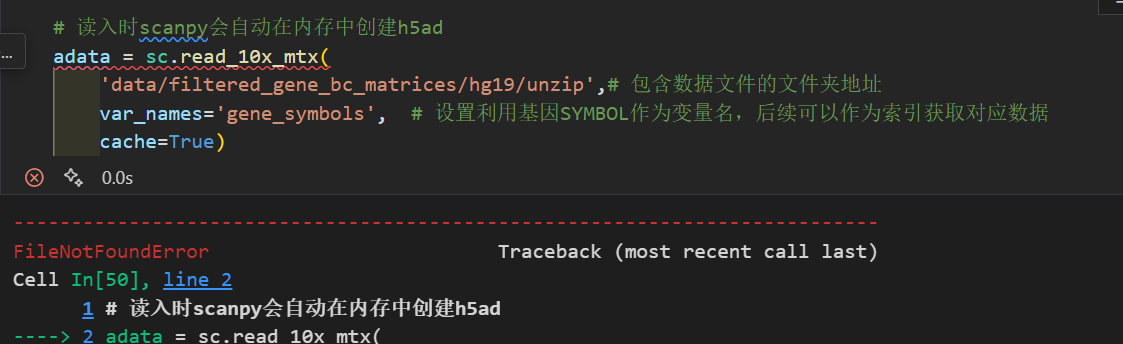
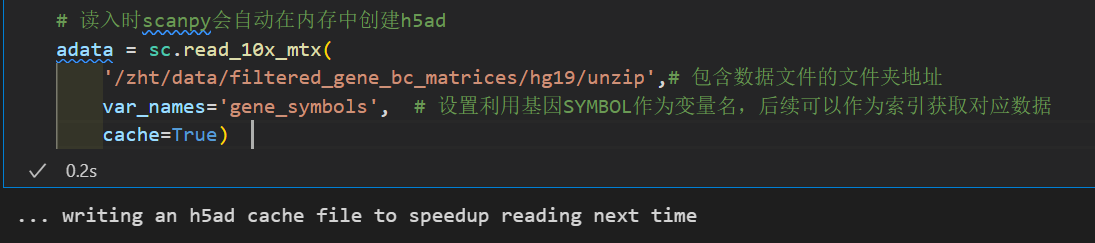
接下来那我们就可以利用scanpy进行读取并创建scanpy的分析对象AnnData。别担心,我们并不需要像上面演示的那样一行一行的去读取:
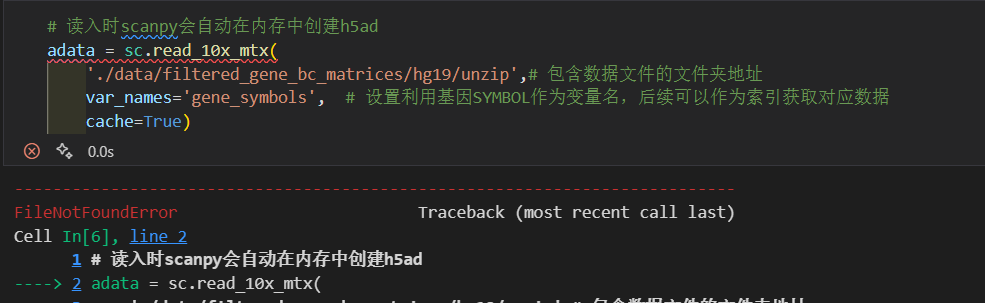
# 读入时scanpy会自动在内存中创建h5ad
adata = sc.read_10x_mtx(
'data/filtered_gene_bc_matrices/hg19/',# 包含数据文件的文件夹地址
var_names='gene_symbols', # 设置利用基因SYMBOL作为变量名,后续可以作为索引获取对应数据
cache=True)
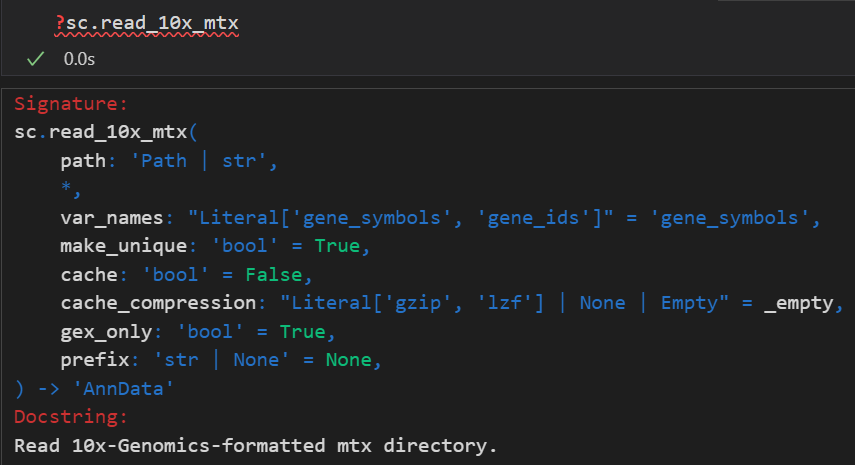
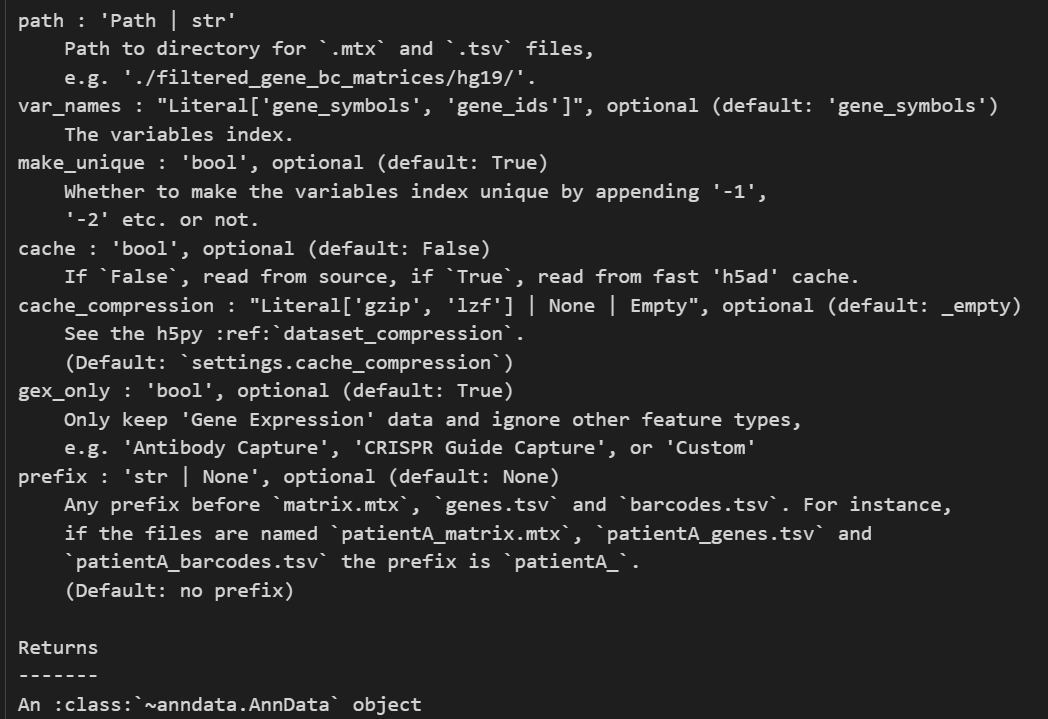
注意,这里提供的文件地址是包含cellranger输出的3个标准文件的地址
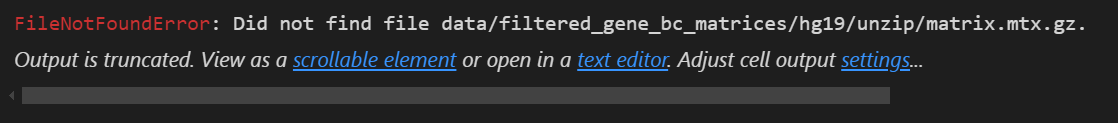
当然如果是直接读取非gzip文件的话,比如说
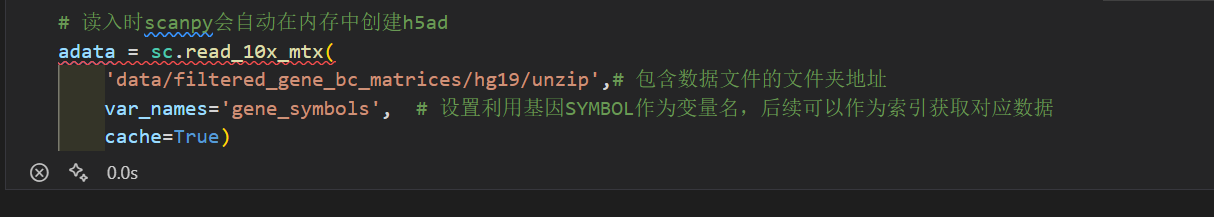
# 读入时scanpy会自动在内存中创建h5ad
adata = sc.read_10x_mtx(
'data/filtered_gene_bc_matrices/hg19/',# 包含数据文件的文件夹地址
var_names='gene_symbols', # 设置利用基因SYMBOL作为变量名,后续可以作为索引获取对应数据
cache=True)

类似的问题可以参考seurat版本的要求:
https://mp.weixin.qq.com/s/AwiH57D6U83baHnro00sGQ
https://mp.weixin.qq.com/s/AjkY_uUdCBjwg7wgUiZ-cg
其实就是函数版本的需求
查看具体函数:

其实按理来说提供了
总之暂时将所有的文件进行压缩gzip处理
查看github上的issue:https://github.com/scverse/scanpy/issues/3457
同样的问题,还是与时俱进
只能说从结果上去看,is_legacy也就是传统分析并没有奏效,
**那如果我们所有的文件效果都是在is_legacy是false的情况下分析的话,我们有必要将10x的经典3个文件修改如下:
**
**也就是:
**都gzip,genes需要更改为features
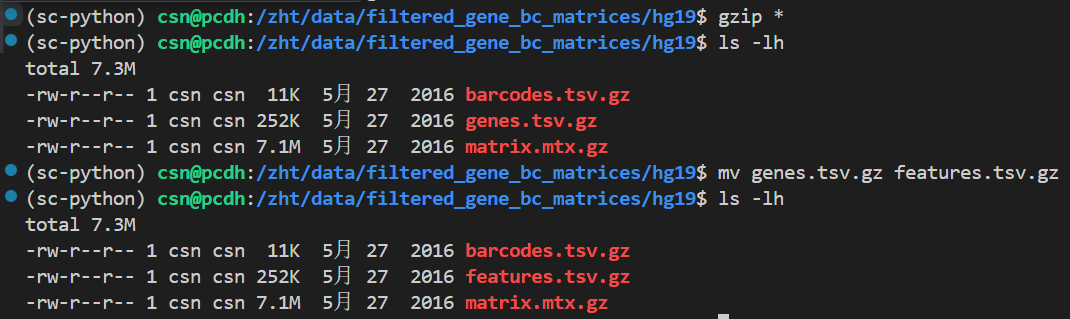
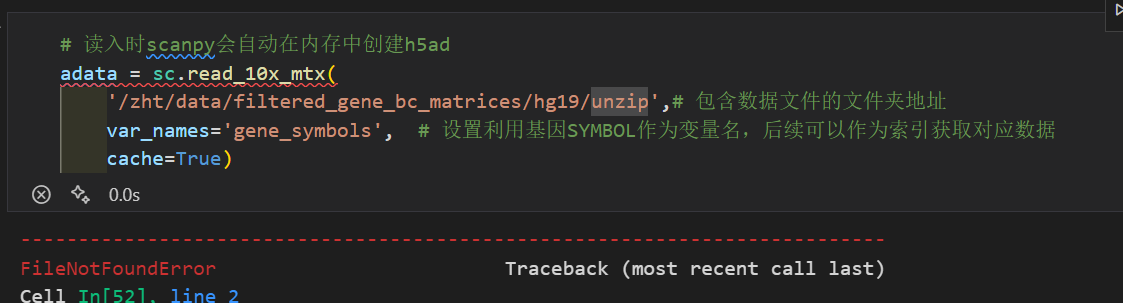
但是还是会报错,需要绝对路径
# 读入时scanpy会自动在内存中创建h5ad
adata = sc.read_10x_mtx(
'/zht/data/filtered_gene_bc_matrices/hg19/',# 包含数据文件的文件夹地址
var_names='gene_symbols', # 设置利用基因SYMBOL作为变量名,后续可以作为索引获取对应数据
cache=True)
新的报错:
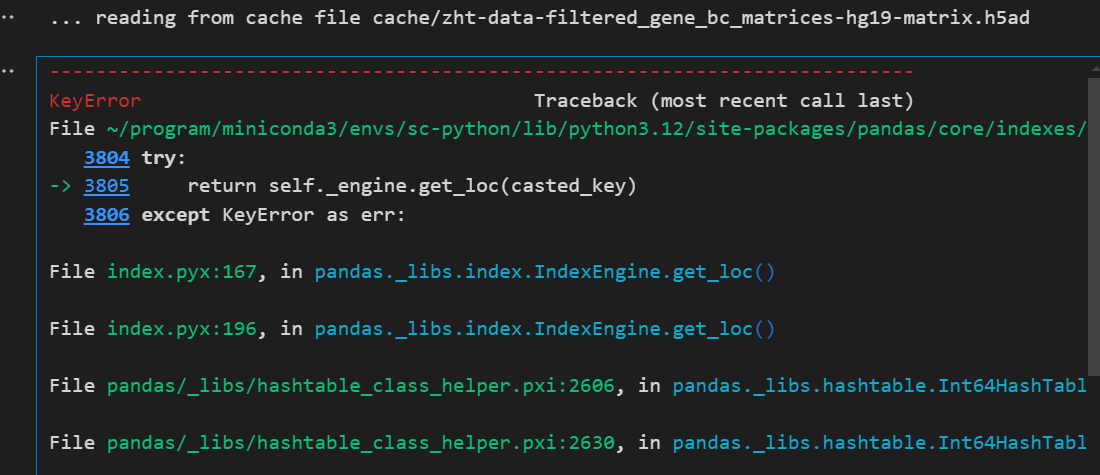
https://github.com/scverse/scanpy/issues/1916
文件格式也没问题:
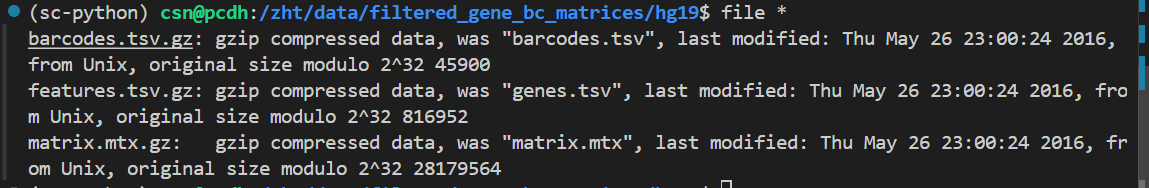
参考:
https://github.com/scverse/scanpy/issues/2053
有另外一个办法处理
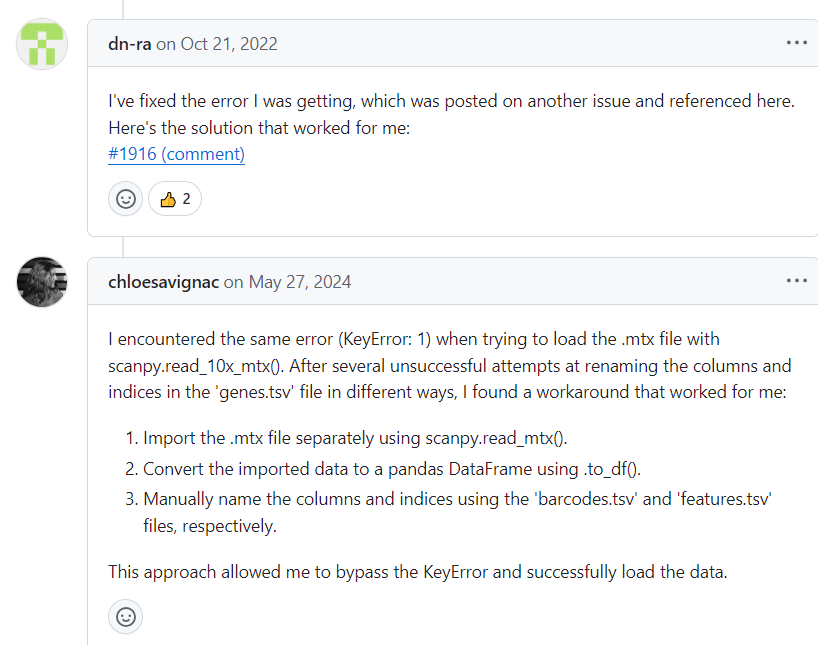
实际上参考的就是前面的https://github.com/scverse/scanpy/issues/1916
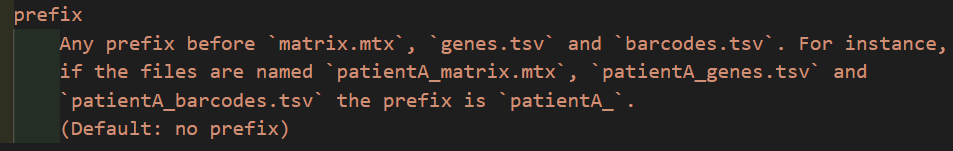
虽然从理论上讲,貌似只需要matrix.mtx,genes.tsv,barcodes.tsv文件即可
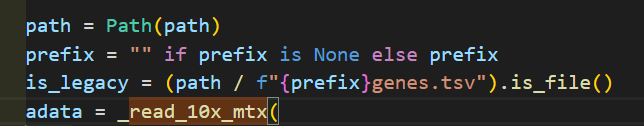
问题可不可能是前面使用相对路径、绝对路径差异上,导致此处函数未能识别出is_legacy的bool值,然后我又修改成了gz文件
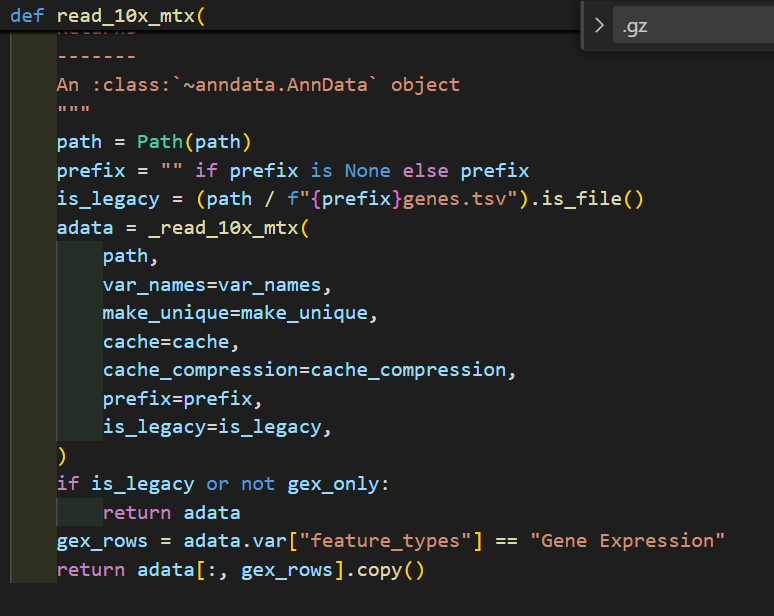
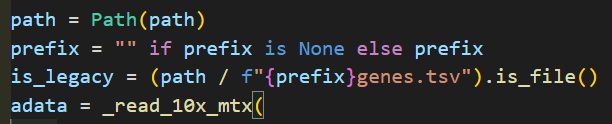
首先我使用v3的未gz版本:
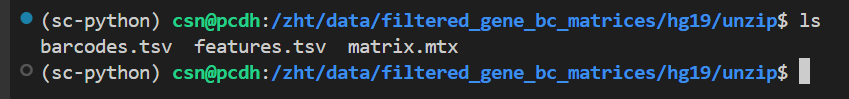
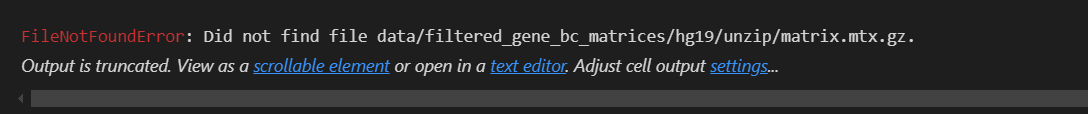
报错:
如果我使用绝对路径:还是同样报错

如果我将features转换为genes,也就是v2版本
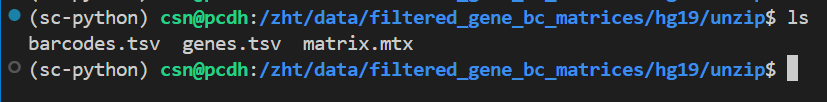
结果就阅读成功了!
如果我不是用绝对路径,而是自以为的相对路径:
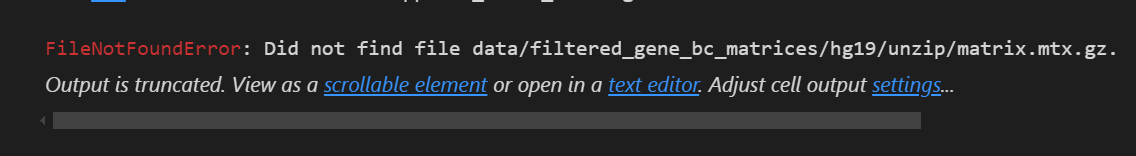
就会报错!同样的报错,所以随时使用绝对路径
所以问题其实很简单!还是那个逻辑,我们先不考虑seurat中的处理差异
至少对于scanpy来说,我们需要关注cellranger version处理版本的差异!那么差异体现在哪里呢?
首先v2是legacy经典处理方式:
我们需要确保的是genes.tsv,而不是features.tsv,另外这个文件也不能有后缀,即gz
总体来说:
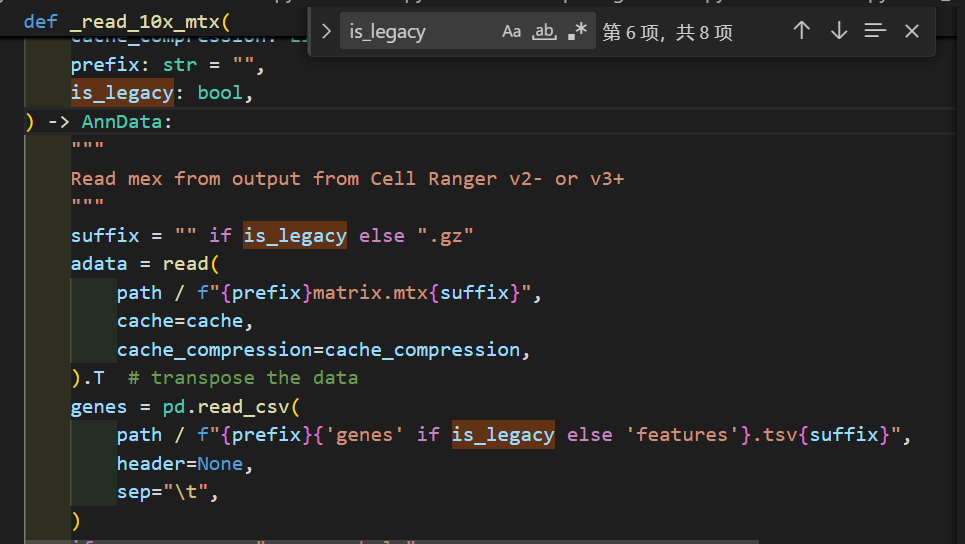
函数read_10x_mtx定义的过程中会调用_read_10x_mtx函数,
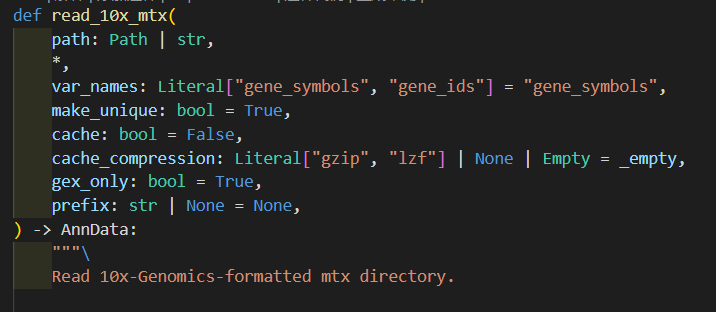
然后判断是否是v2 legacy经典分析的条件:
我们可以看到在调用_read_10x_mtx前就已经先判断了一次,就是看目录中有无genes.tsv文件
如果有,就是经典分析,如果没有就不是经典v2
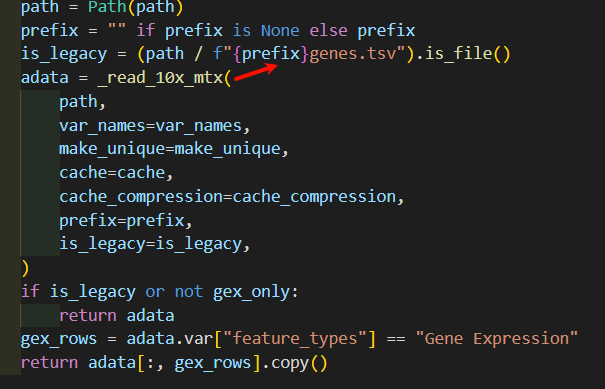
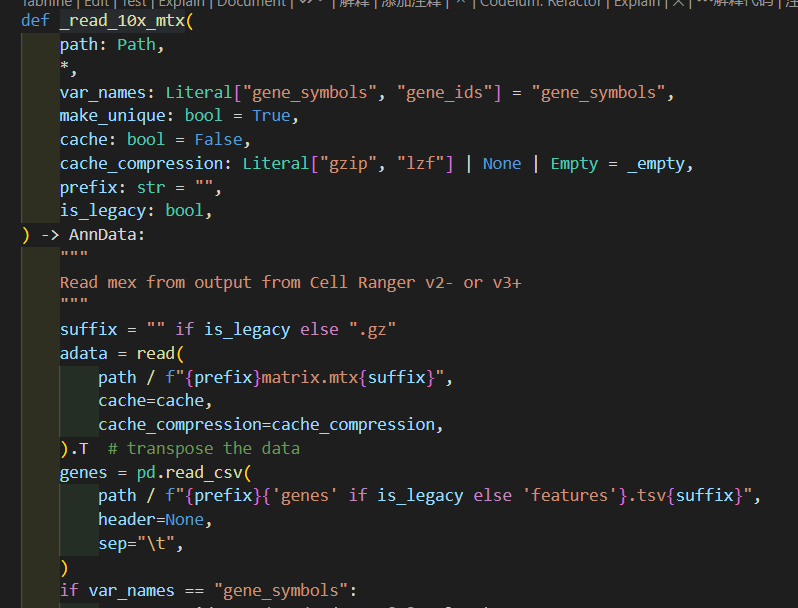
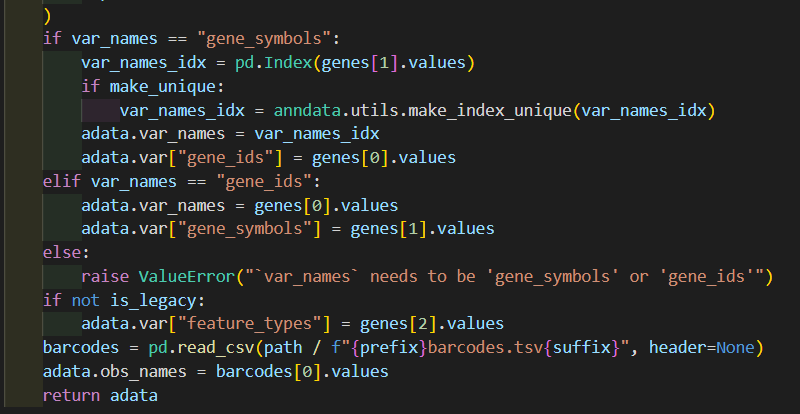
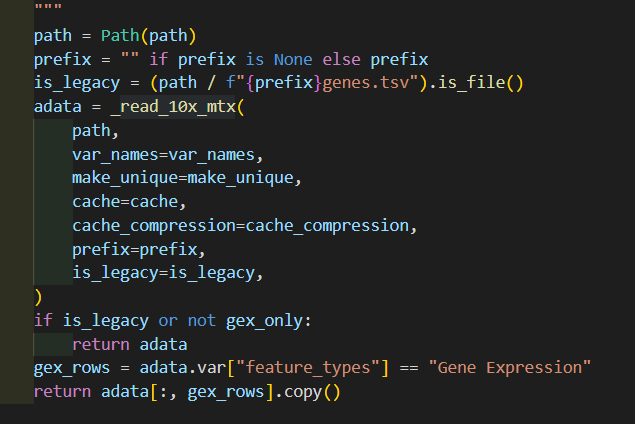
然后深入_read_10x_mtx这个函数,其实我们可以看到里面并没有对is_legacy值进行二次判断赋值,

然后里面是对后缀进行了分析,如果是经典v2的话,就没有gz后缀,suffix为空,否则v3非经典就是gz
然后后面的文件读取实际上就是都朝着is_lagacy以及suffix的这两个条件判断进行的组合分析:
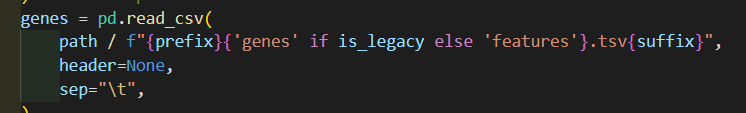
genes如果是经典的话就是genes.tsv,否者是v3的features.tsv.gz
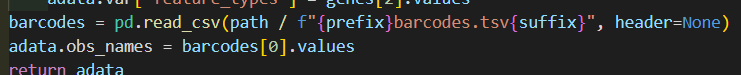
barcodes同理
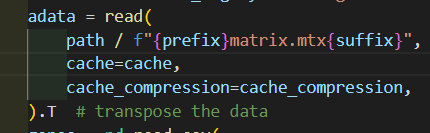
matrix矩阵同理
——》
综上:文件读取格式非常严格
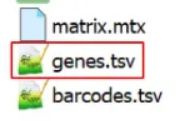
v2/legacy在scanpy中:
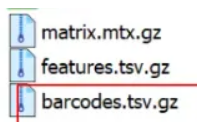
v3/非legacy在scanpy中:
理论上这应该是cellranger处理的时候就应该明确的处理前提,
那么实际处理公共数据库的过程中也会有人上传v2的然后更名features,并且进行gz压缩,需要注意!
那这样处理的话我们该如何识别呢?
比如说我们前面处理的3个文件应该是v2 legacy的,我如果上传的时候神不知鬼不觉地更名了并gz压缩:
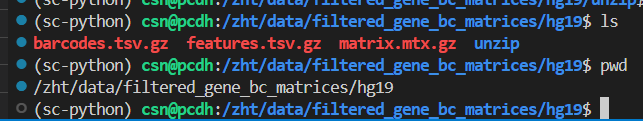
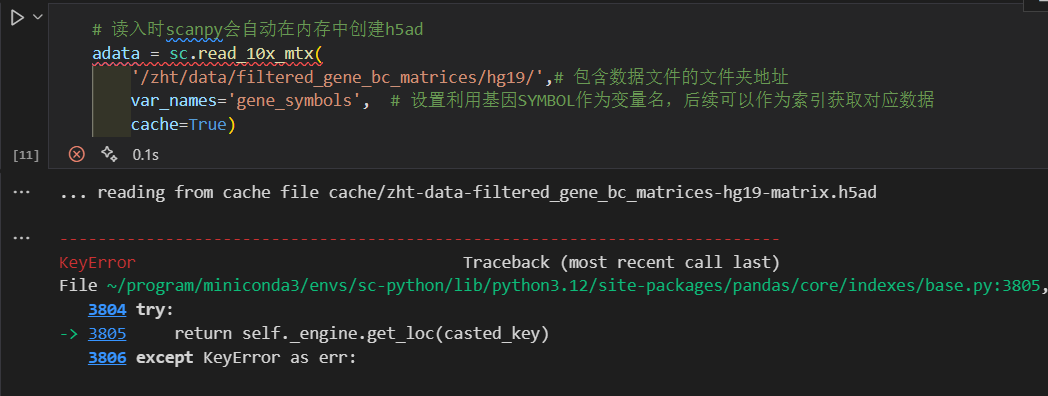
可以发现会报错!
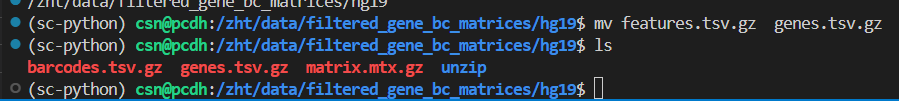
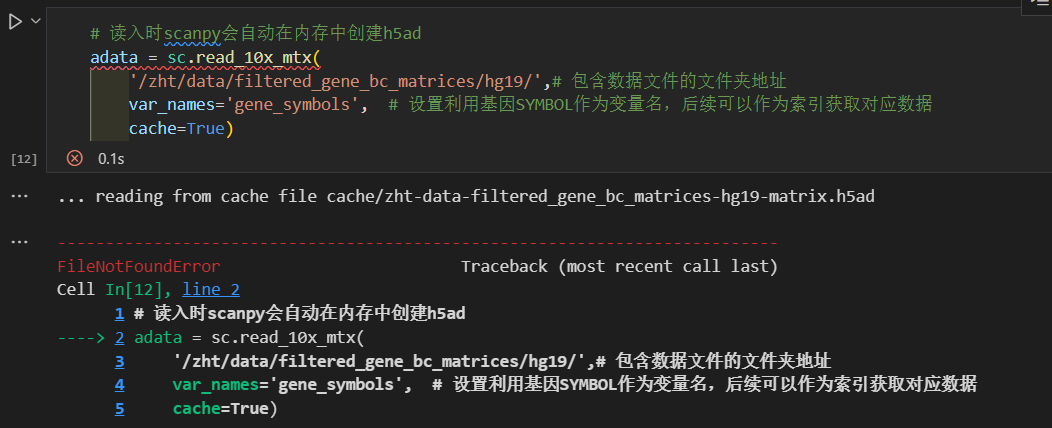
就算是不完全的更名的trick也会被报错识别出来!
所以对于一个没有源头的数据,如何确定该如何读入,因为数据格式具有迷惑性,我们不能只是看数据后缀以及命名怎么样?
唯一的办法:
就是分别按照前面v2以及v3的格式修改文件,然后试一试两种读取方式,哪种没报错,说明文件就是哪种处理过来的,从报错结果倒推cellranger版本;

综上此步:
# 读入时scanpy会自动在内存中创建h5ad
adata = sc.read_10x_mtx(
'/zht/data/filtered_gene_bc_matrices/hg19/',# 包含数据文件的文件夹地址
var_names='gene_symbols', # 设置利用基因SYMBOL作为变量名,后续可以作为索引获取对应数据
cache=True)
然后我们一般处理的话,对于gene还是用symbol多,投稿的时候,画图的时候,分析的时候展示最多的是symbol;
所以就是分析需要后面的gene是symbol,做过bulk RNA-seq分析的都知道,gene id有很多种;
symbol是少得可怜,基本上就是蛋白编码gene那一块,一般转换的时候无法一一对应上Ensembl的gene id,因为转录本之类的id会比较复杂;
但是我们这里genes是两列,所以需要对id列对应的symbol列去重
# #如果使用var_names="gene_symbols"这一参数加载数据,可能会有重复的基因名字出现,因为一个symbol是有可能对应不止一个gene id的
#这时候就需要下面这句代码,这句代码会添加额外字符来使基因名唯一化
# 在读取数据时候,若选择var_names='gene_ids',则不需要此步控制.var唯一
adata.var_names_make_unique()
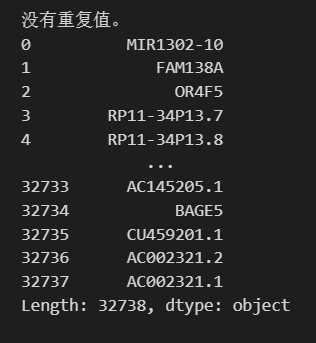
其实我们可以判断是否去重,使用pandas
import pandas as pd
# 将 adata.var_names 转换为 pandas Series
var_names_series = pd.Series(adata.var_names)
# 检查是否存在重复值
has_duplicates = var_names_series.duplicated().any()
if has_duplicates:
print("存在重复值。")
# 打印重复值
duplicates = var_names_series[var_names_series.duplicated()]
print("重复值如下:")
print(duplicates)
else:
print("没有重复值。")
print(var_names_series)
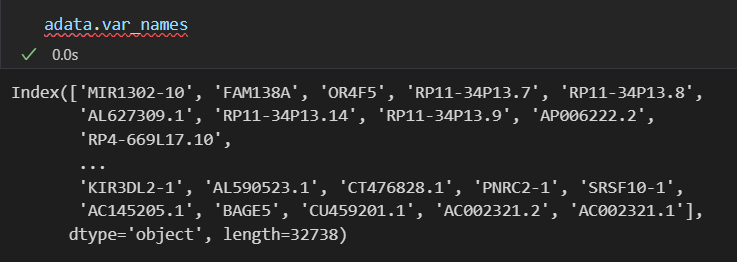
当然,这个例子比较特殊,我们还是建议时刻加上这句代码(同样如果我们使用gene id,虽然有可能不会重复,但是也要加上)
总之,都加上这句
实际读取的时候还可以读入其他格式的数据:
(4)数据处理

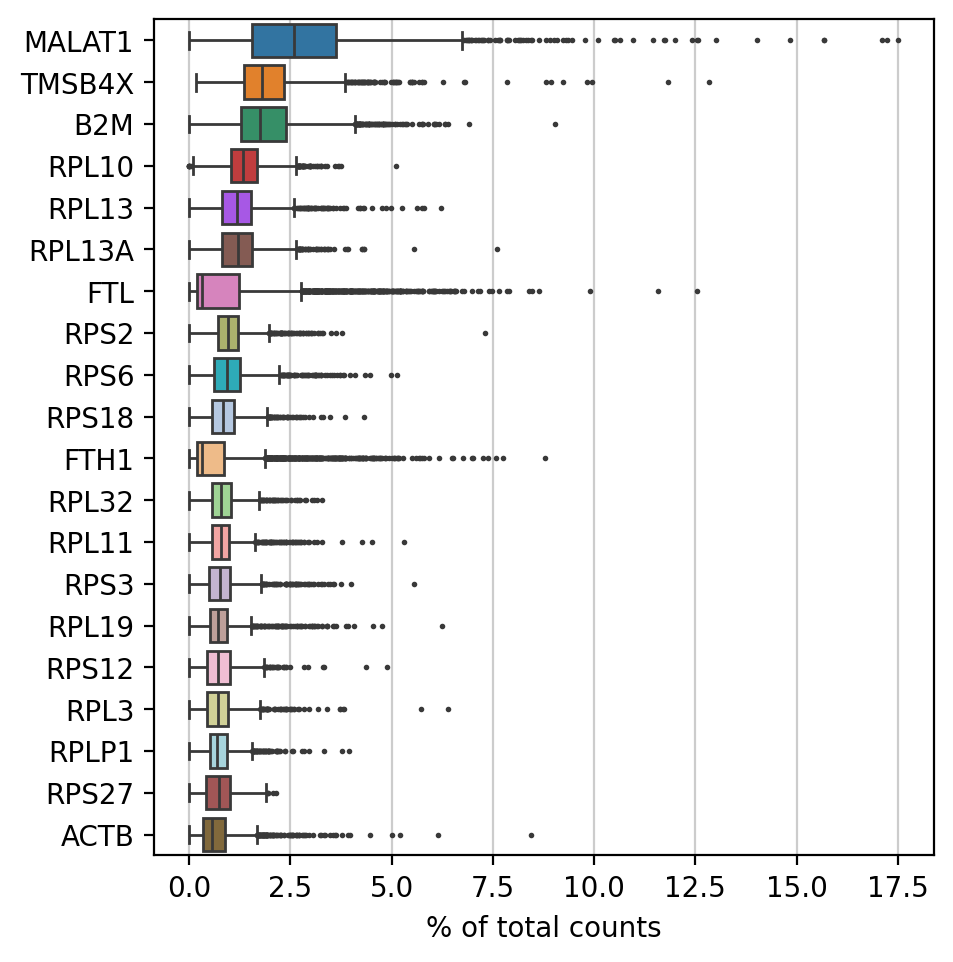
# 计算并以箱线图的形式展示表达量最高的二十个基因:
#绘制每个基因在所有细胞中的计数分数的图形,n_top参数表示要显示的基因数量
#统计基因在细胞中的占比并可视化
#每一个gene在一个cell中占据所有cell的表达量有一个值,类似于reads,或者是counts,或者是bulk中的TPM等值,总之这个值可以计算比例;然后我们计算所有细胞中这个gene的比例,那就是总共有cell数目个比例,再然后就是绘制箱线图,也就是每一个基因在所有细胞中的平均表达量(这里计算了百分比含量)
sc.pl.highest_expr_genes(adata, n_top=20, )

- 数据结构:
- 在
AnnData对象中,行表示细胞,列表示基因。
- 在
- 计算比例:
- 对于每个基因,该函数计算在每个细胞中分配给该基因的总计数的比例。这意味着,对于每个细胞,你需要确定该基因的读取数占该细胞总读取数的多少。
- 聚合结果:
- 在为每个细胞和基因计算完这些比例后,该函数会计算每个基因在所有细胞中的平均比例。
- 箱线图可视化:
- 然后,它识别出平均比例最高的前
n_top个基因,并创建箱线图以可视化这些比例在所有细胞中的分布。
- 然后,它识别出平均比例最高的前
简单来说:你有一个矩阵,其中行是细胞,列是基因,而该函数计算、平均并可视化了基因表达作为每个细胞总计数比例的分布。箱线图有助于说明这些顶级基因在不同细胞中的表达情况,展示变异、中位数和潜在的离群值。
# 过滤掉表达基因数量不足两百的细胞:
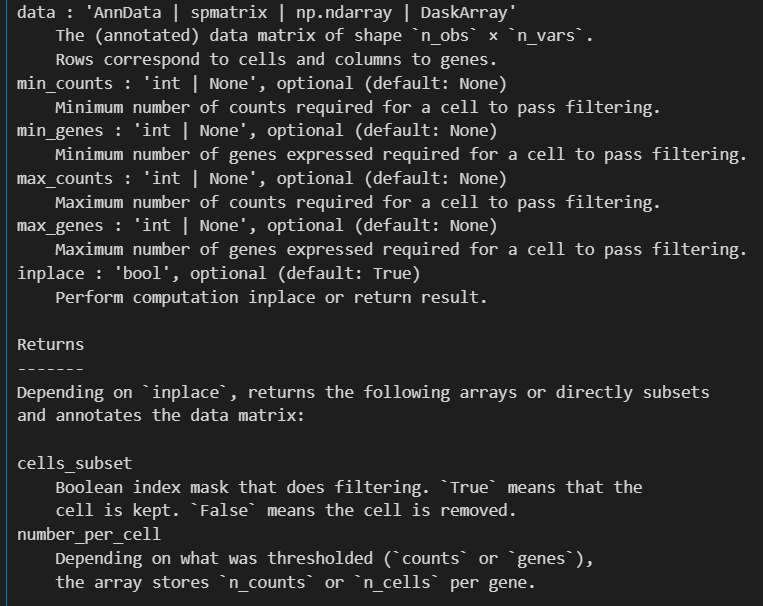
sc.pp.filter_cells(adata, min_genes=200)
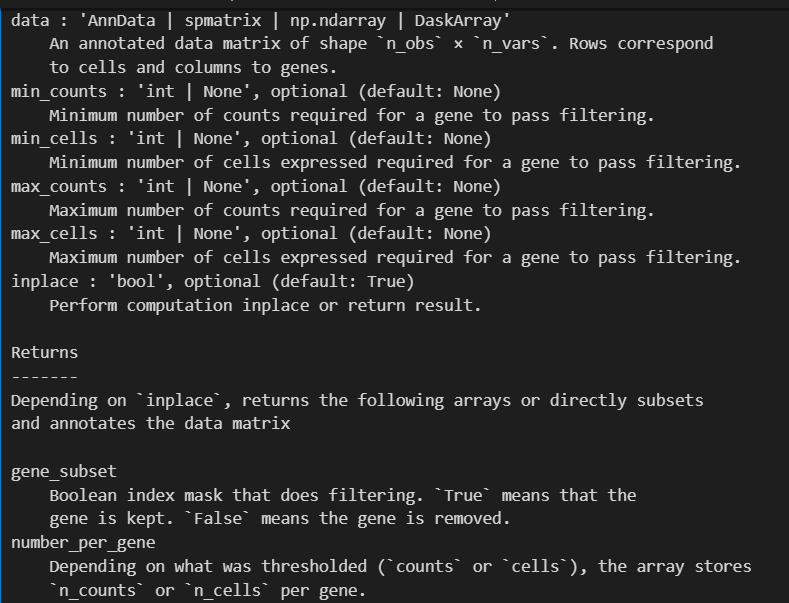
# 过滤掉表达细胞数量不足3个的基因:
sc.pp.filter_genes(adata, min_cells=3)
## filtered out 19024 genes that are detected in less than 3 cells
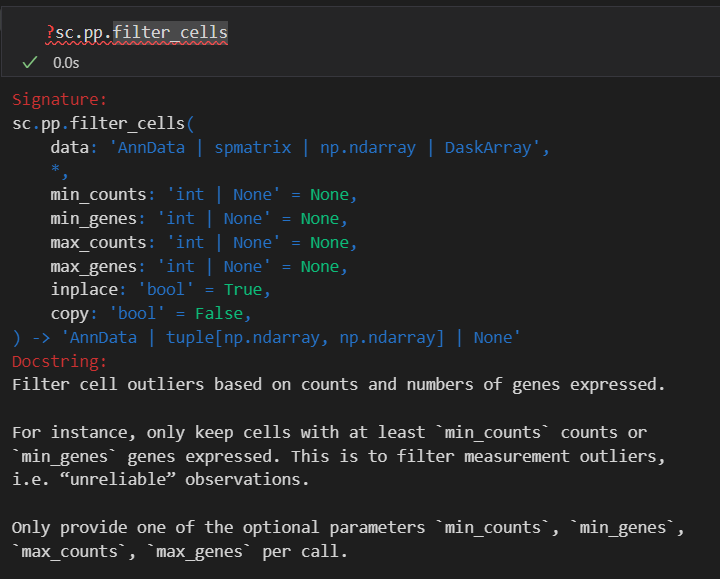
就是过滤掉gene低表达量(该cell中所有的gene表达总量加起来)的cell
同样过滤掉低表达量cell数目的gene(该gene在所有的cell中表达的数目过少)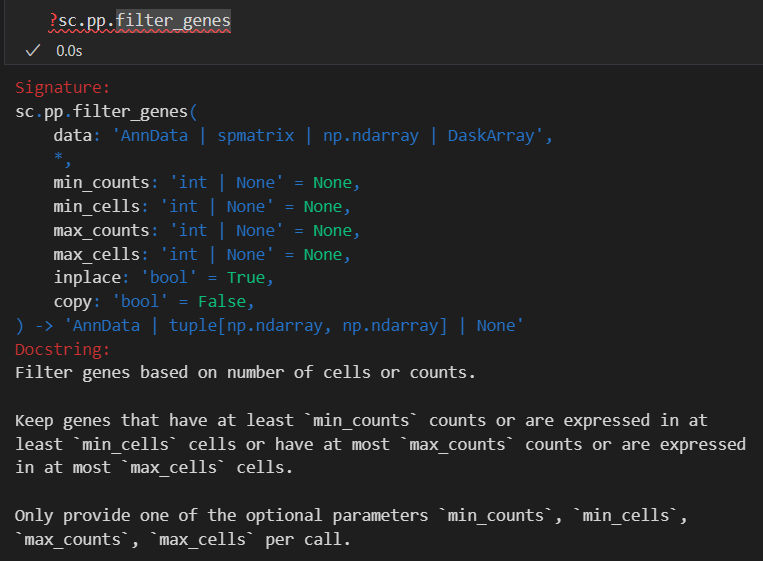
现在再来看这个过滤之后的数据:
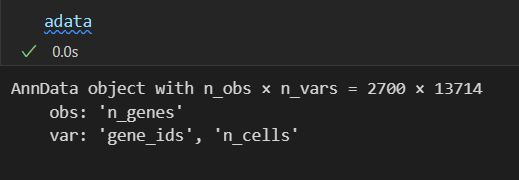
本来是2700x32738
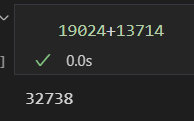
就是过滤之后的数据+被过滤掉的数据
总之过滤掉低gene表达量的cell,以及低cell表达量的gene,
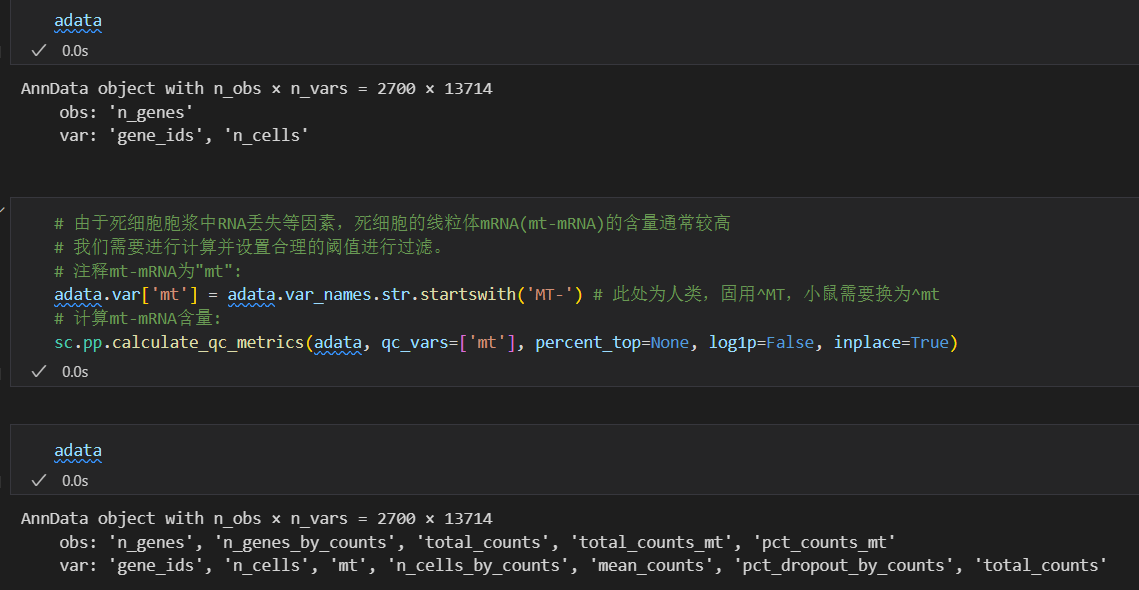
接着就是过滤掉死细胞,注意是过滤cell,然后死细胞的话有个指标就是mt-mRNA含量的问题,
# 由于死细胞胞浆中RNA丢失等因素,死细胞的线粒体mRNA(mt-mRNA)的含量通常较高
# 我们需要进行计算并设置合理的阈值进行过滤。
# 注释mt-mRNA为"mt":
adata.var['mt'] = adata.var_names.str.startswith('MT-') # 此处为人类,固用^MT,小鼠需要换为^mt
# 计算mt-mRNA含量:
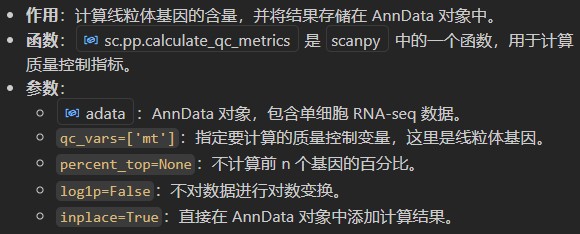
sc.pp.calculate_qc_metrics(adata, qc_vars=['mt'], percent_top=None, log1p=False, inplace=True)
其实我们前面这么多gene,3w多个本身就是包含有线粒体gene了
新增1列用于存放线粒体gene名字
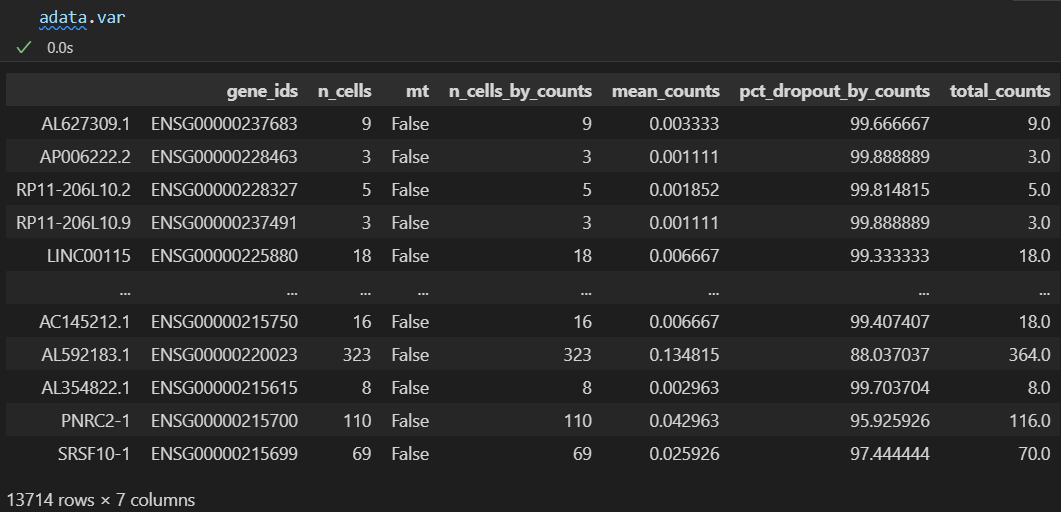
此时再看我们的数据:
obs和var上都有了很多属性
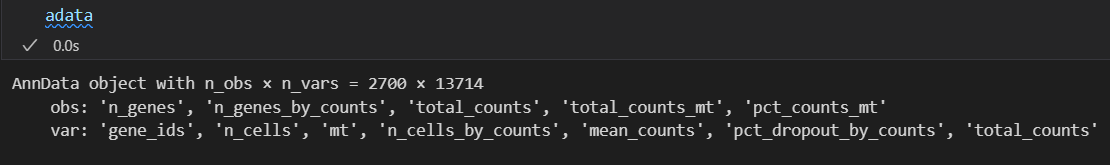
对比之下都是上面函数处理获得的
主要是注意上面的expr_type以及var_type等
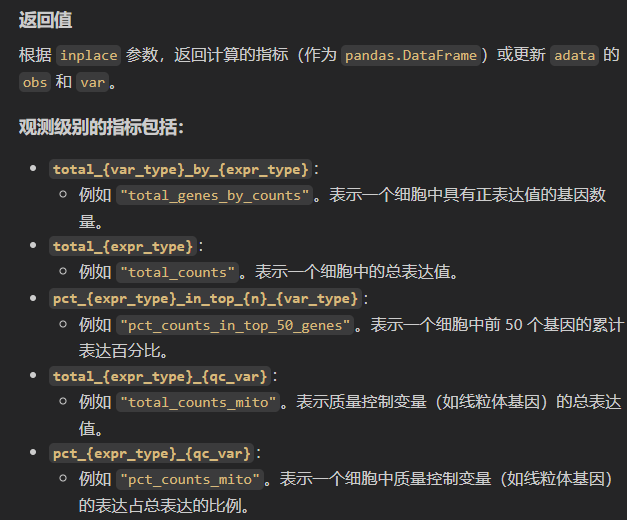
这些多出来的列名说明与示例
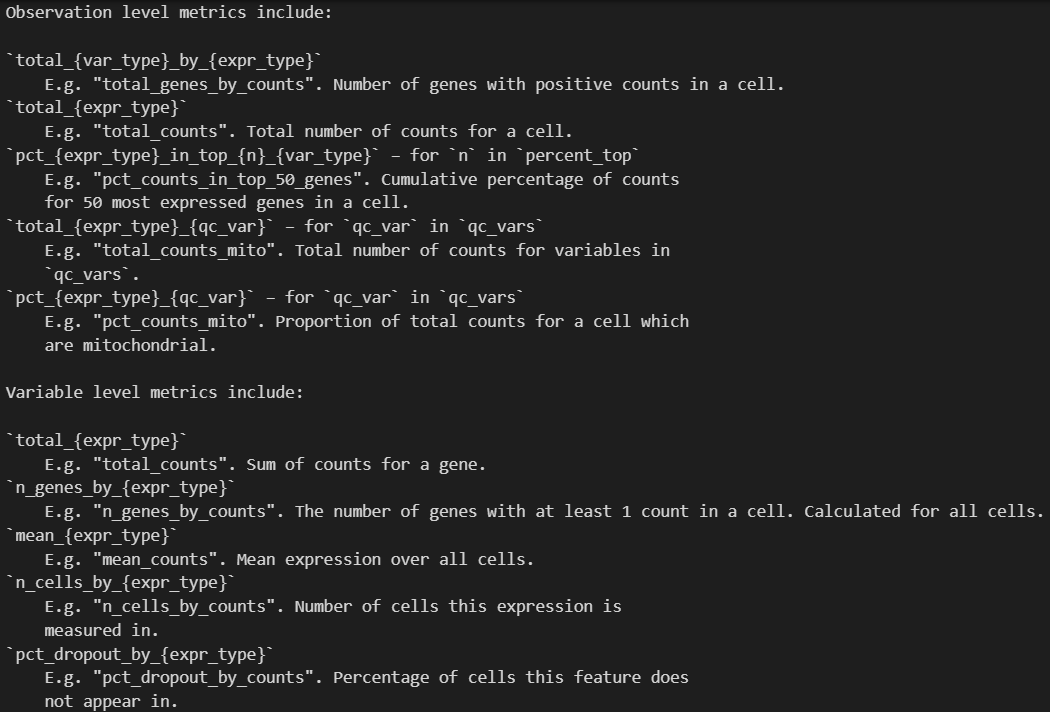
1. expr_type
- 作用:定义表达值的类型(如
"counts"、"log1p"),影响生成的指标名称中的{expr_type}占位符。 - 示例:
sc.pp.calculate_qc_metrics(adata, expr_type="counts")
- 生成的指标名如 `"total_counts"`(总计数)和 `"mean_counts"`(平均计数)。
- 若设为 `expr_type="log1p"`,则指标名变为 `"total_log1p"` 或 `"mean_log1p"`。
2. var_type
- 作用:定义变量类型(默认
"genes"),影响{var_type}占位符的替换。 - 示例:
sc.pp.calculate_qc_metrics(adata, var_type="peaks")
- 观测指标中的 `"total_genes_by_counts"` 变为 `"total_peaks_by_counts"`。
- 变量指标中的 `"n_genes_by_counts"` 变为 `"n_peaks_by_counts"`。
3. qc_vars
- 作用:指定需要额外计算的质控变量(如线粒体基因、核糖体基因)。
- 示例:
sc.pp.calculate_qc_metrics(adata, qc_vars=["mito", "ribo"])
- 生成观测指标:
* `"total_counts_mito"`(线粒体基因总计数)和 `"total_counts_ribo"`(核糖体基因总计数)。
* `"pct_counts_mito"`(线粒体基因占比)和 `"pct_counts_ribo"`(核糖体基因占比)。
4. percent_top
- 作用:指定计算前
n个高表达基因的累计百分比。 - 示例:
sc.pp.calculate_qc_metrics(adata, percent_top=[50, 100])
- 生成观测指标:
* `"pct_counts_in_top_50_genes"`(前50个基因的计数占比)。
* `"pct_counts_in_top_100_genes"`(前100个基因的计数占比)。
5. inplace
- 作用:决定是否直接更新
adata或返回 DataFrame。 - 示例:
# 返回两个 DataFrame:obs_metrics 和 var_metrics
obs_metrics, var_metrics = sc.pp.calculate_qc_metrics(adata, inplace=False)
# 直接更新 adata.obs 和 adata.var
sc.pp.calculate_qc_metrics(adata, inplace=True)
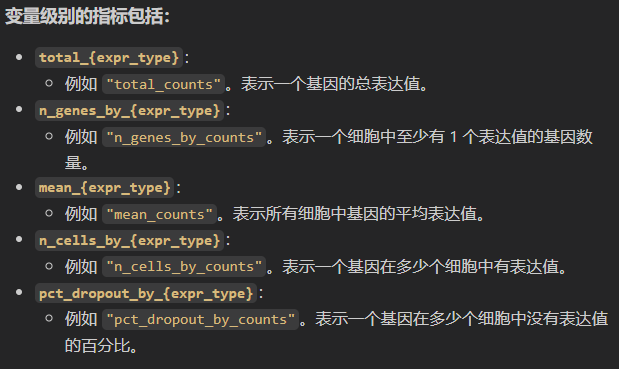
返回的指标解释与示例
假设输入数据如下:
- 细胞总数:
1000个。 - 基因总数:
20000个。 - 线粒体基因占比:
5%。
观测级别(细胞)指标示例:
| 指标名称 | 示例值 | 说明 |
|---|---|---|
total_genes_by_counts | 1500 | 某细胞中表达量 >0 的基因数。 |
total_counts | 10000 | 某细胞总计数(UMI数)。 |
pct_counts_in_top_50_genes | 30% | 前50个高表达基因的计数占比。 |
total_counts_mito | 500 | 某细胞中线粒体基因的总计数。 |
pct_counts_mito | 5% | 线粒体基因占该细胞总计数的比例。 |
变量级别(基因)指标示例:
| 指标名称 | 示例值 | 说明 |
|---|---|---|
total_counts | 1,000 | 某基因在所有细胞中的总计数。 |
n_genes_by_counts | 800 | 表达该基因的细胞数。 |
mean_counts | 0.5 | 该基因在所有细胞中的平均计数。 |
n_cells_by_counts | 800 | 表达该基因的细胞数。 |
pct_dropout_by_counts | 20% | 未检测到该基因的细胞比例。 |
参数与指标的关联总结
| 参数 | 影响的指标名称模式 | 示例参数值 | 生成的指标示例 |
|---|---|---|---|
expr_type | total_{expr_type}, mean_{expr_type} | expr_type="counts" | total_counts, mean_counts |
var_type | total_{var_type}_by_{expr_type} | var_type="peaks" | total_peaks_by_counts |
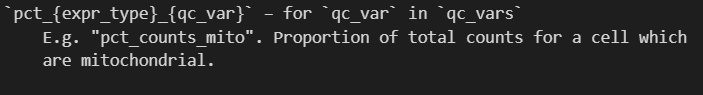
qc_vars | total_{expr_type}_{qc_var} | qc_vars=["mito"] | total_counts_mito |
percent_top | pct_{expr_type}_in_top_{n}_{var_type} | percent_top=[50] | pct_counts_in_top_50_genes |
函数概述
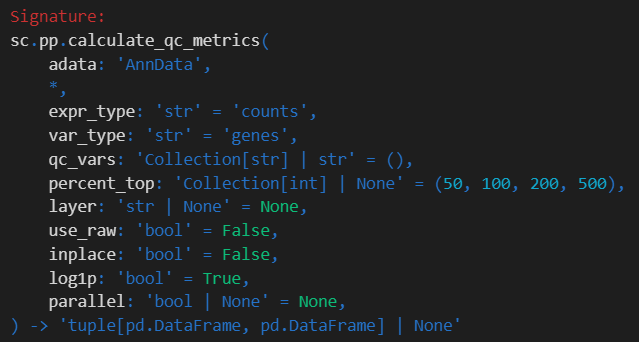
c.pp.calculate_qc_metrics 函数用于计算单细胞 RNA 测序(scRNA-seq)数据的质量控制(QC)指标。这些指标有助于评估数据质量,通过提供基因表达水平、线粒体基因含量以及其他相关统计信息来实现。
参数解释
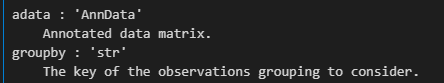
**adata**- 目的: 函数使用的主要数据结构。它应该是一个包含基因表达矩阵和相关元数据的 AnnData 对象。
- 示例:
- 假设您有一个名为
adata的 AnnData 对象,其中包含来自 1,000 个细胞和 2,500 个基因的数据。
- 假设您有一个名为
**qc_vars**- 目的: 用于计算特定 QC 指标的变量(基因)标识符列表。这些通常是基因的子集,例如线粒体基因,它们对于评估细胞活力非常重要。
- 示例:
- 如果您想关注线粒体基因,可以将
qc_vars设置为['mt']。这告诉函数计算诸如线粒体基因的总计数(total_counts_mito)和线粒体基因的计数百分比(pct_counts_mito)等指标。
- 如果您想关注线粒体基因,可以将
**percent_top**- 目的: 确定在计算累积百分比指标时要考虑的高表达基因数量。这有助于了解每个细胞中基因表达的分布情况。
- 示例:
- 如果将
percent_top设置为[50],函数将计算诸如pct_counts_in_top_50_genes等指标,该指标表示每个细胞中前 50 个高表达基因的累积计数百分比。 - 数值示例: 在一个总共有 10,000 个计数的细胞中,如果前 50 个基因贡献了 7,500 个计数,那么
pct_counts_in_top_50_genes将为 75%。
- 如果将
**log1p**- 目的: 一个布尔值标志,用于指示在计算 QC 指标之前是否对数据应用对数转换(
log1p,即log(1 + x))。对数转换可以稳定方差并使数据更接近正态分布。 - 示例:
- 将
log1p设置为False表示不应用任何转换。所有计算均使用原始计数。 - 将
log1p设置为True将在计算指标之前对数据应用log1p转换。 - 数值示例:
- 对于原始计数 100,如果
log1p=True,转换后的值将是log(1 + 100) ≈ 4.615。
- 对于原始计数 100,如果
- 将
- 目的: 一个布尔值标志,用于指示在计算 QC 指标之前是否对数据应用对数转换(
**inplace**- 目的: 确定计算的 QC 指标是直接添加到
adata对象的.obs(观测/细胞级别)和.var(变量/基因级别)属性中,还是作为单独的 DataFrame 返回。 - 示例:
- 将
inplace设置为True将通过向adata.obs(例如total_counts、pct_counts_mito)和adata.var(例如每个基因的total_counts)添加新列来修改adata对象。 - 将
inplace设置为False将返回一个包含计算指标的pandas.DataFrame,而不会修改原始adata对象。 - 数值示例:
- 如果
inplace=True,在运行函数后,访问adata.obs['total_counts']可能会返回每个细胞的值,例如[5000, 7500, 6200, ...]。
- 如果
- 将
- 目的: 确定计算的 QC 指标是直接添加到
- 观测级别指标:
**total_genes_by_counts**- 描述: 细胞中具有正计数的基因数量。
- 示例: 一个细胞表达了 1,200 个基因,总共有 2,500 个基因。
**total_counts**- 描述: 每个细胞的总计数(UMIs 或 reads)。
- 示例: 一个细胞的总计数为 10,000。
**pct_counts_in_top_50_genes**- 描述: 前 50 个高表达基因的累积计数百分比。
- 示例: 75% 的计数来自前 50 个基因。
**total_counts_mito**- 描述: 线粒体基因的总计数。
- 示例: 一个细胞中有 1,500 个线粒体计数。
**pct_counts_mito**- 描述: 总计数中线粒体计数的百分比。
- 示例: 15% 的计数是线粒体的。
- 变量级别指标:
**n_genes_by_counts**- 描述: 每个细胞中至少有 1 个计数的基因数量。
- 示例: 基因 X 在 1,000 个细胞中的 800 个细胞中被检测到。
**mean_counts**- 描述: 每个基因在所有细胞中的平均表达(平均计数)。
- 示例: 基因 Y 的平均计数为每个细胞 50 个。
**n_cells_by_counts**- 描述: 表达某个基因的细胞数量。
- 示例: 基因 Z 在 1,000 个细胞中的 950 个细胞中被表达。
**pct_dropout_by_counts**- 描述: 某个基因未被表达的细胞百分比。
- 示例: 基因 W 的 5% 未被表达,即在 1,000 个细胞中的 50 个细胞中未被表达。
实际示例
import scanpy as sc
# 假设 'adata' 是您的 AnnData 对象,包含 1,000 个细胞和 2,500 个基因的数据
adata = sc.datasets.pbmc3k() # 示例数据集
# 注释线粒体基因(假设为人类;对于小鼠使用 'mt')
adata.var['mt'] = adata.var_names.str.startswith('MT-')
# 计算 QC 指标
sc.pp.calculate_qc_metrics(
adata,
qc_vars=['mt'], # 线粒体基因
percent_top=50, # 前 50 个基因
log1p=False, # 不进行对数转换
inplace=True # 将指标添加到 'adata'
)
# 访问计算的指标
print(adata.obs[['total_counts', 'total_genes', 'pct_counts_mt', 'pct_counts_in_top_50_genes']].head())
输出示例:
total_counts total_genes pct_counts_mt pct_counts_in_top_50_genes
0 5000 1200 15.0 75.0
1 7500 1500 10.0 80.0
2 6200 1100 12.0 70.0
3 8000 1300 8.0 85.0
4 4500 900 20.0 65.0
在这个示例中:
**total_counts**显示每个细胞的总计数。**total_genes**表示每个细胞中表达的基因数量。**pct_counts_mt**表示线粒体基因的计数百分比。**pct_counts_in_top_50_genes**显示每个细胞中前 50 个高表达基因的累积计数百分比。
通过理解并适当设置 c.pp.calculate_qc_metrics 函数中的每个参数,可以有效评估您的单细胞 RNA 测序数据的质量。数值示例有助于解释这些指标,确保数据预处理和下游分析基于可靠且高质量的数据。
总之这个qc评估函数可以统计很多指标,
然后我们可以看一下自己的数据的obs以及var对象变化如何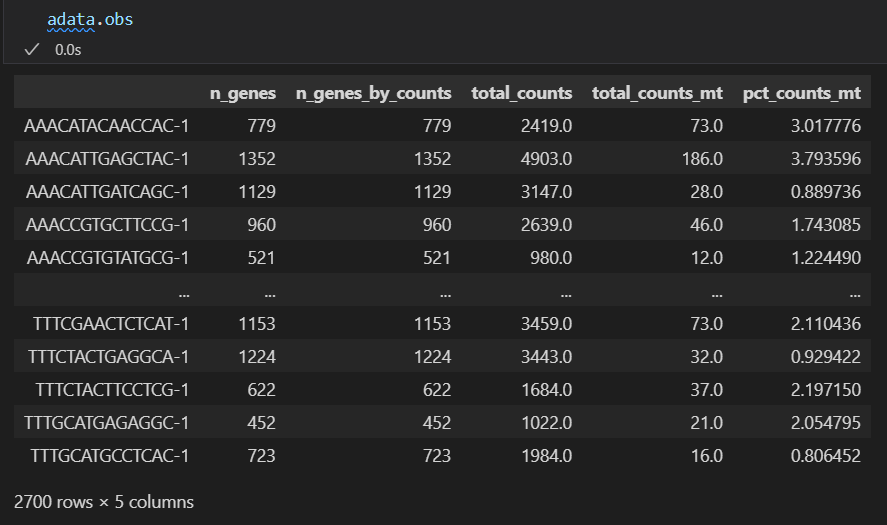
然后这些指标可以进行可视化,主要是箱线图、小提琴图之类:
# 可视化n_genes_by_counts、total_counts、pct_counts_mt等质控指标
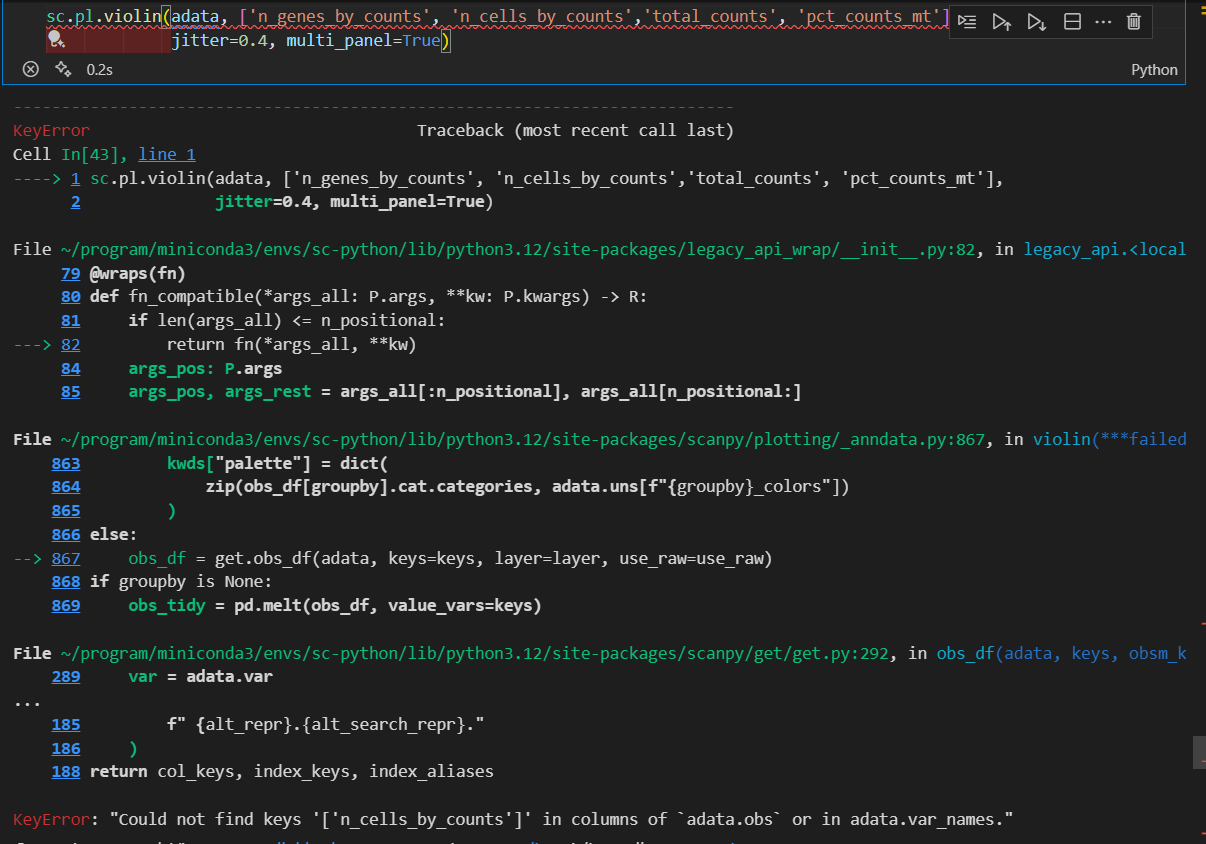
sc.pl.violin(adata, ['n_genes_by_counts', 'total_counts', 'pct_counts_mt'],
jitter=0.4, multi_panel=True)
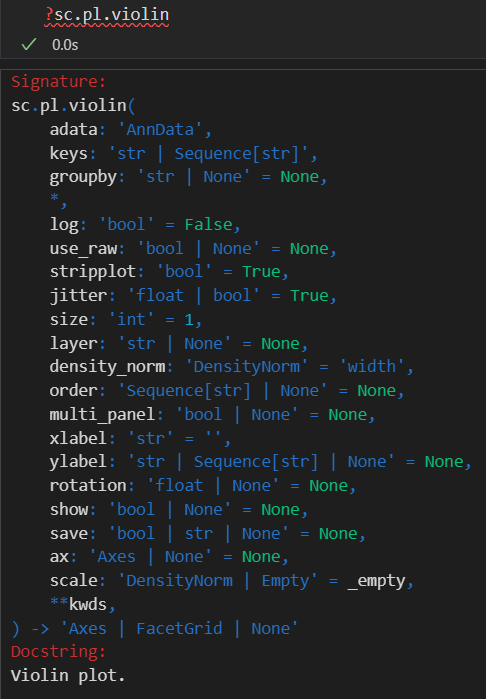
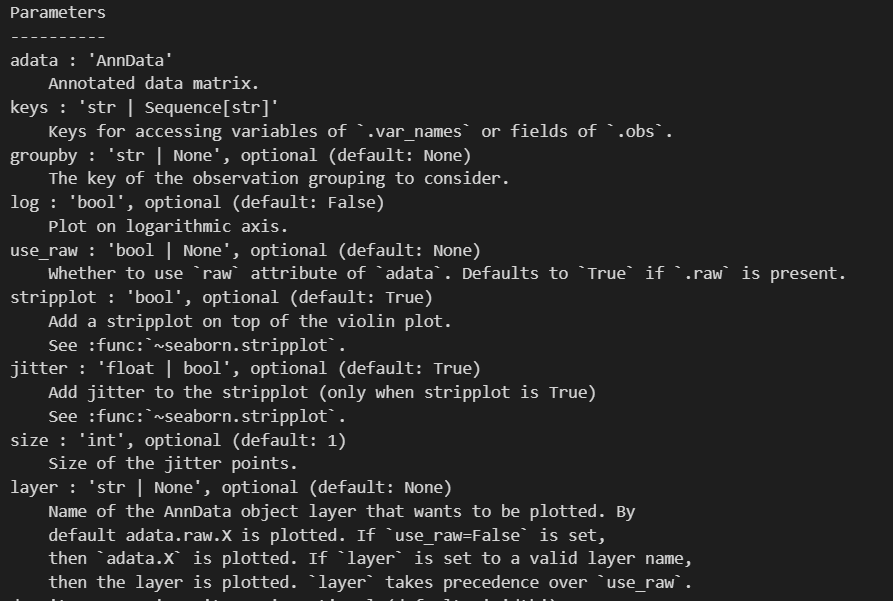
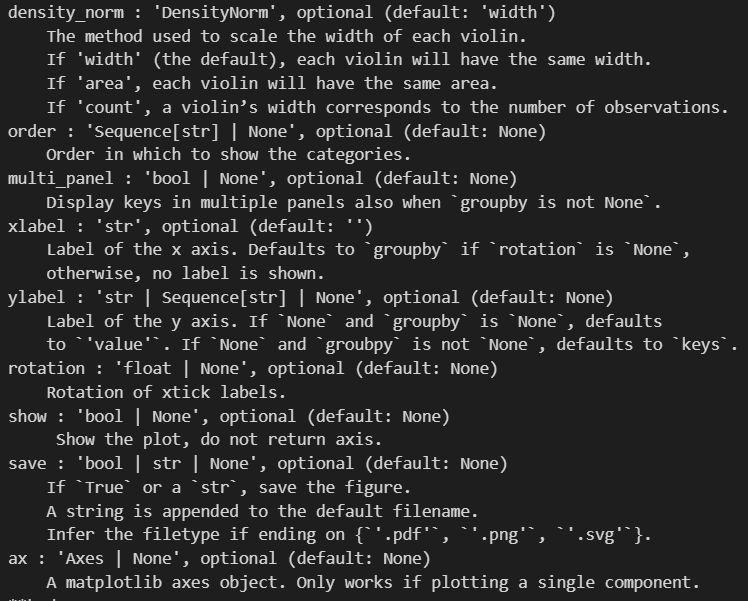
生成的三张小提琴图代表:表达基因的数量,每个细胞包含的表达量,线粒体基因表达量的百分比
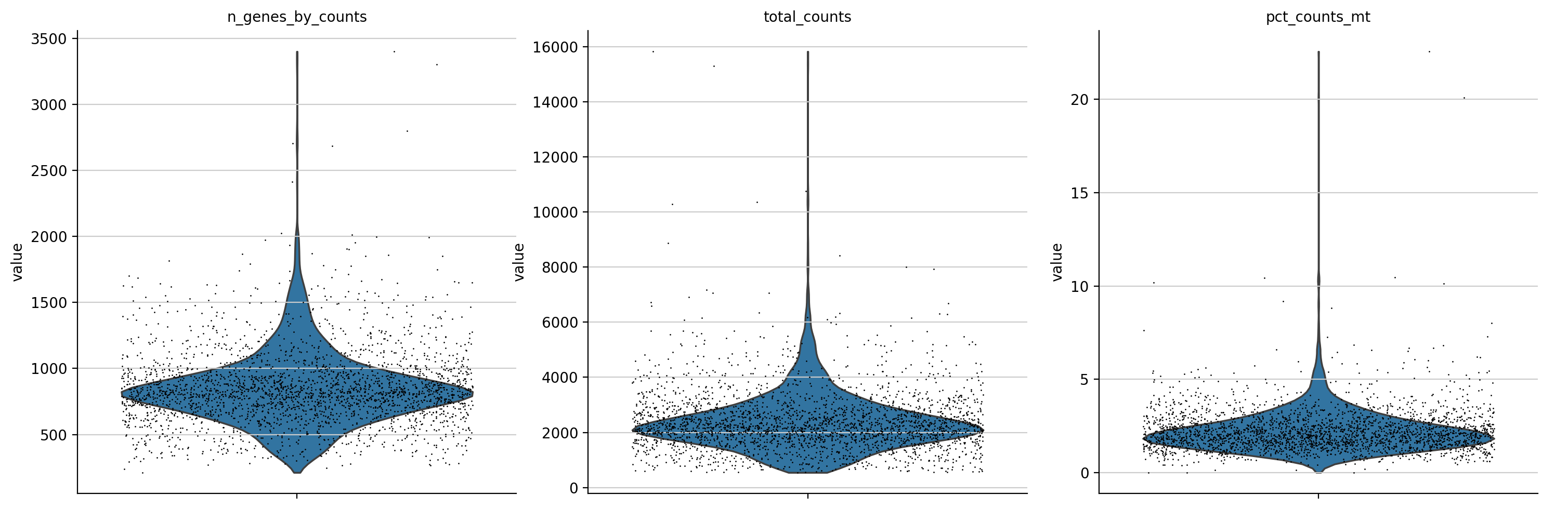
上面展示的指标:


每1个小点jitter都是1个cell,然后就是筛选出来在该cell中表达的gene数目(说起来是gene的数目,实际上只要某个gene有1个count落在这个cell中,就可以说是这个gene在该cell中表达,然后是计算所有的cell),可以看得出来大部分cell大概有1k左右个gene在表达。(in a cell,其实就是for a cell)


同样的,每一个小点都是一个cell,然后计算的是这个cell中所有的gene的count的总和(for a cell),注意和var中的同名指标区别

每一个小点都是一个cell,然后这个cell里面是线粒体gene的counts占据所有的counts的比例(for a cell)
看的出来大部分的cell,其对应的mt DNA的比例都很低。
其实只要查看这个指标是在obs还是在var中,就能够理解是for a cell(obs的指标),还是for a gene(var的指标)。——当然这个方法不是很严谨,只能说除了看指标出现在obs还是var上,最好还是看一下函数帮助文档中的注释!!!!!
其实仔细查看一下这个小提琴的函数:
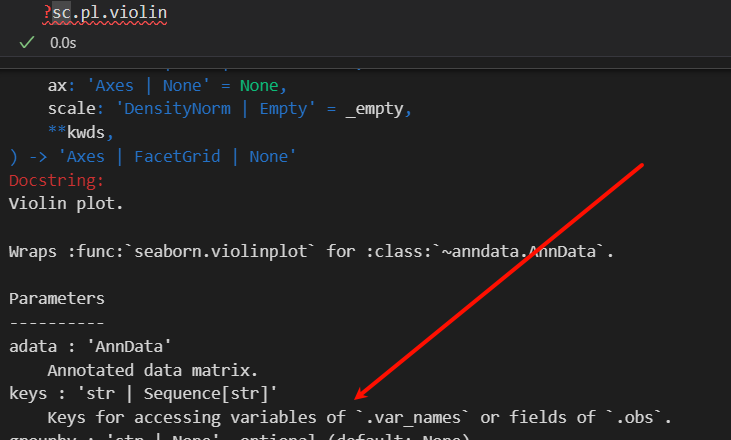
很自然就会想到如何确定展示的列名是obs还是var轴上的,比如说total_counts就有2个同名的变量,那么该如何处理呢?如果我提供了,该如何识别呢?
一个很自然的想法就是规定所有的列名都是在同一个维度轴中的,比如说是obs轴中,或者是var轴中:
见下,应该是选取在同一个obs轴或者是var轴中的
# 展现线粒体count与细胞总count的相关性:
sc.pl.scatter(adata, x='total_counts', y='pct_counts_mt')
依据前面的经验:
其中y轴:每个cell中线粒体 counts 占总 counts 的百分比,纵轴是比例;
x轴:每个cell中所有的gene的count的总和,x轴是数目
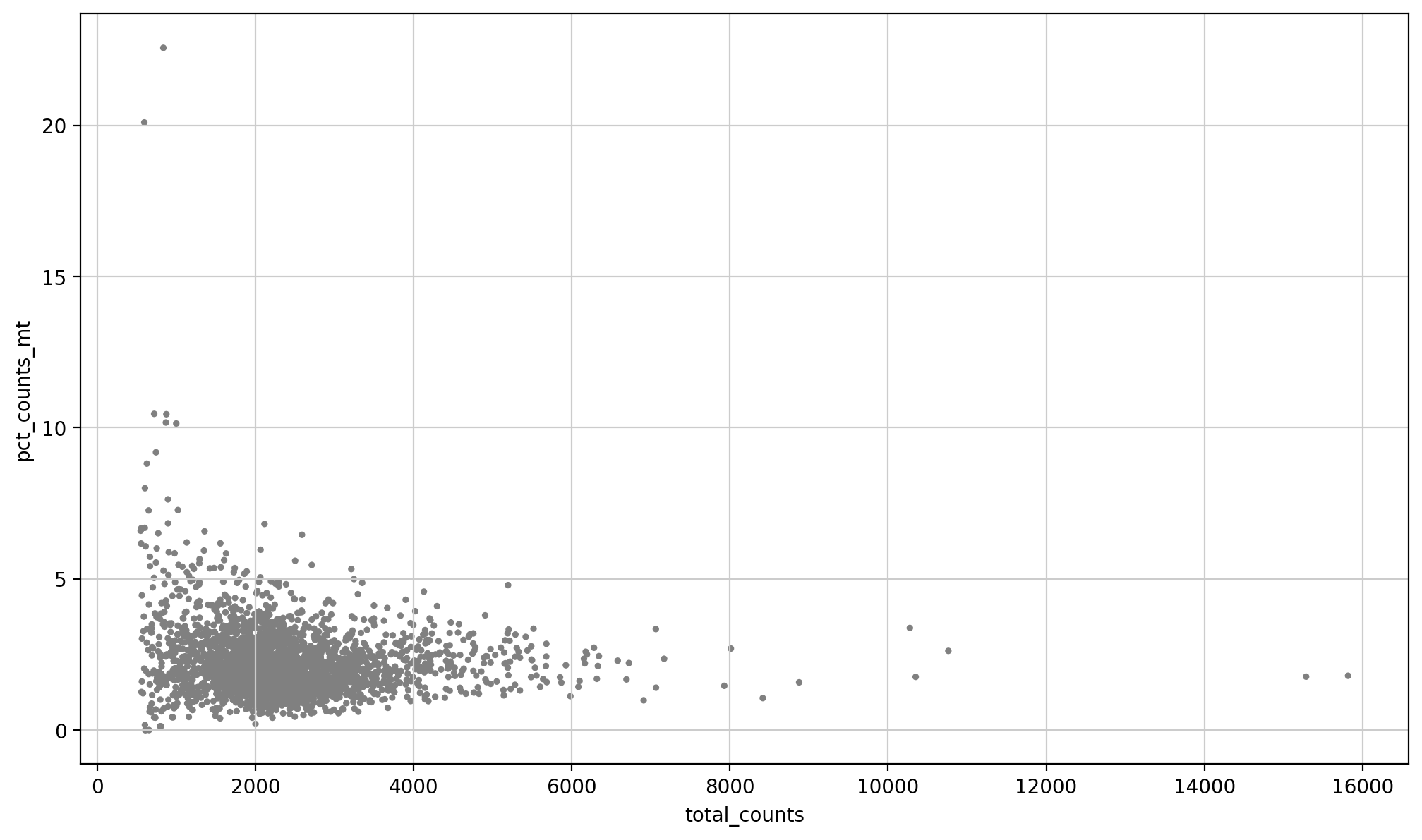
然后注意这里的scatter绘图,要提供的数据是要一一对应的,所以一般是along一个轴即可,比如说是obs轴,或者是var轴;
所以在提供x以及y轴变量的时候,确保同在obs轴上,或者是同在var轴上,然后只需要提供column列名即可;
这里可能就会有人问了,那前面在obs以及var中都看到了total_counts这个指标,那这怎么确定呢?
所以我们只能从适用结果角度上去分析,此处两个使用的都是obs也就是for a cell的指标。
同样的
# 展现细胞基因数量与细胞总count间的相关性
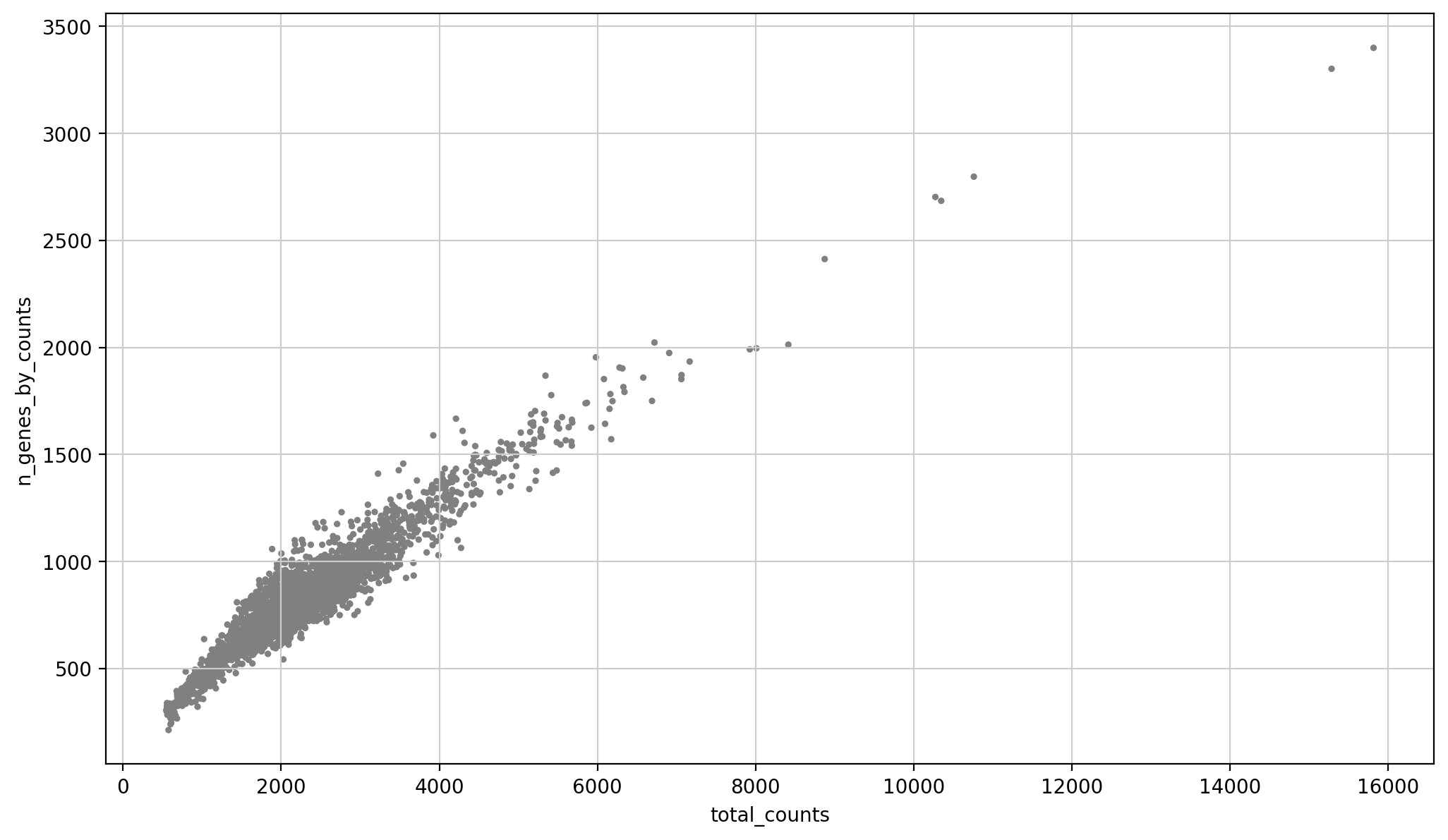
sc.pl.scatter(adata, x='total_counts', y='n_genes_by_counts')
依据前面的经验:
其中y轴:在该cell中表达的gene数目,y轴是数目;
x轴:每个cell中所有的gene的count的总和,x轴是数目
进一步的过滤:
此处的过滤其实是看上面的数据图得出的结论
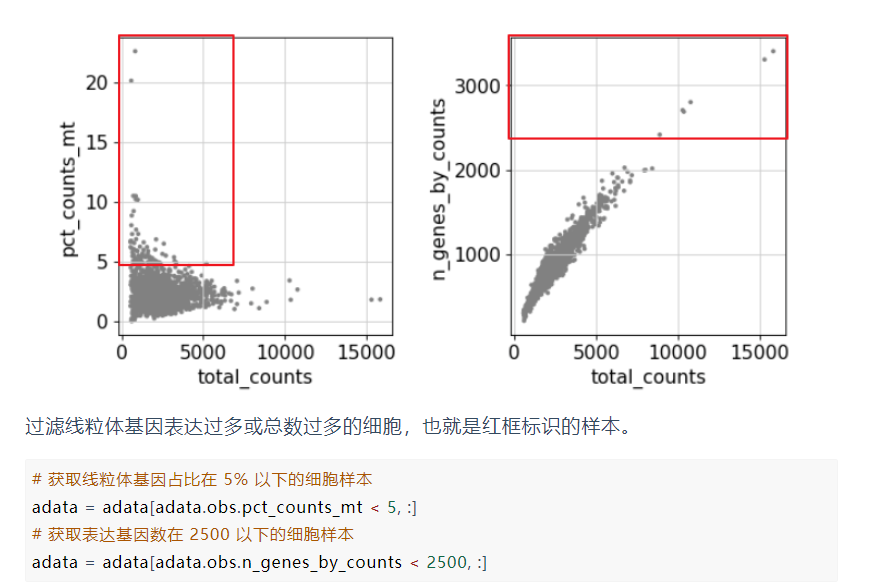
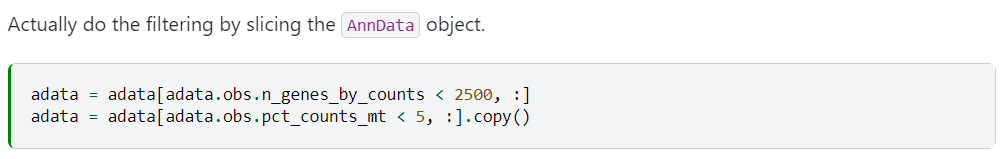
# 保留每个细胞中表达基因少于2500的细胞
adata = adata[adata.obs.n_genes_by_counts < 2500, :]
# 保留细胞中mt-mRNA的counts比例小于5%的细胞
adata = adata[adata.obs.pct_counts_mt < 5, :]
官网此处使用copy
https://scanpy.readthedocs.io/en/stable/tutorials/basics/clustering-2017.html

本质上就是数据切片,都是过滤cell,所以是对obs行数据框对象的切片,使用的是obs的某些列的过滤条件,逻辑本质上就是取切片。
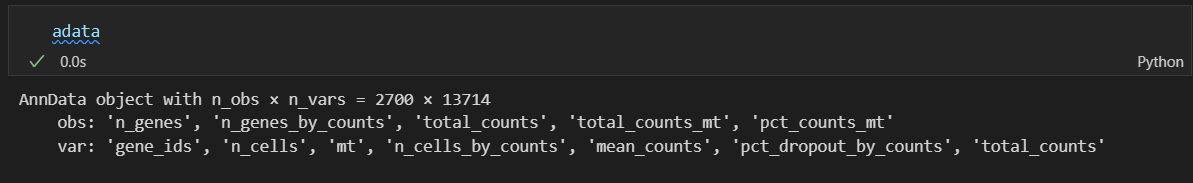
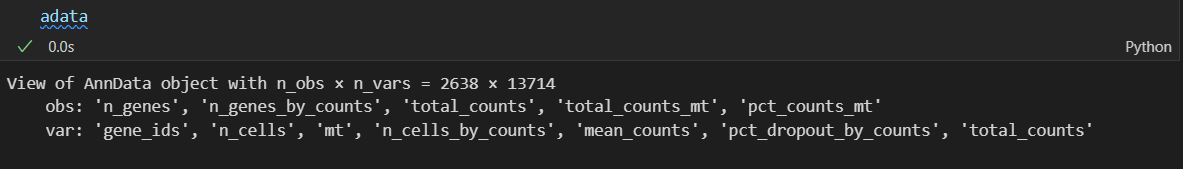
前后筛选其实没有少多少cell
紧接着是进行数据标准化,
# 标准化数据,将每个细胞的reads数标准化至10,000,
# 这使得相同基因在不同细胞之间的表达量比较成为可能
#标准化数据,使每个细胞的总表达量相同(这里设定为10000),消除测序深度的影响。
sc.pp.normalize_total(adata, target_sum=1e4)
一般都是将feature在sample之间进行标准化,使得同一feature可以在不同的sample之间进行比较;
也就是将同一gene的表达值在不同的cell之间进行标准化,使得同1个gene在不同的cell之间的表达值可以进行比较;
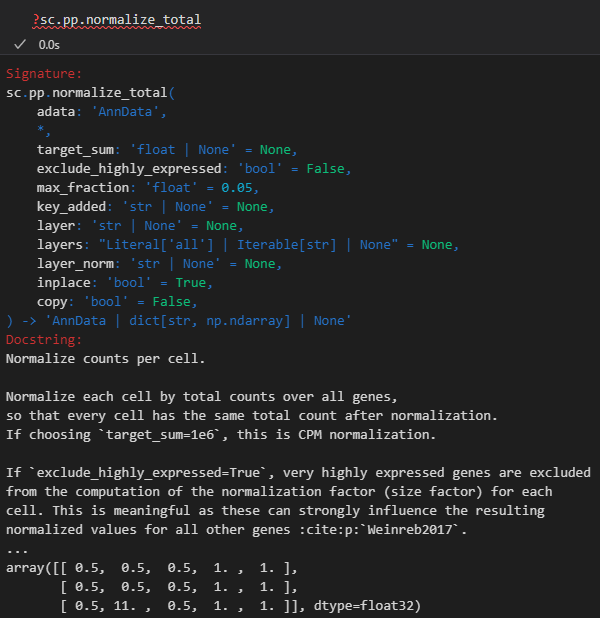
这里是将每个cell中所有gene的counts之和归一化为1e4个
然后再对表达矩阵进行log化操作:
# 对表达矩阵进行log(X+1)的操作
# 对标准化后的数据进行对数转换(log(x+1)),将数据缩放到更容易分析的范围。
sc.pp.log1p(adata)
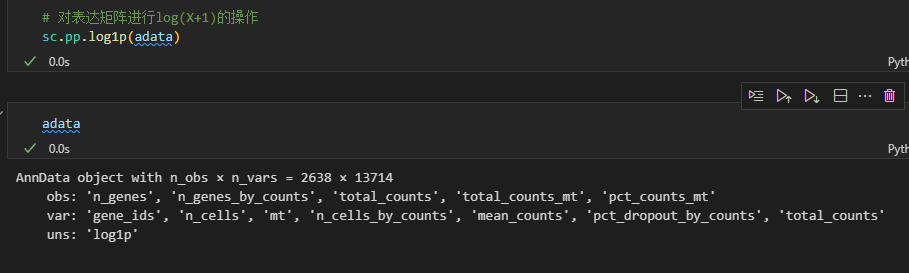
此时在非结构化数据槽中存储了log1p的矩阵:实际上就是自然对数的ln(x+1)
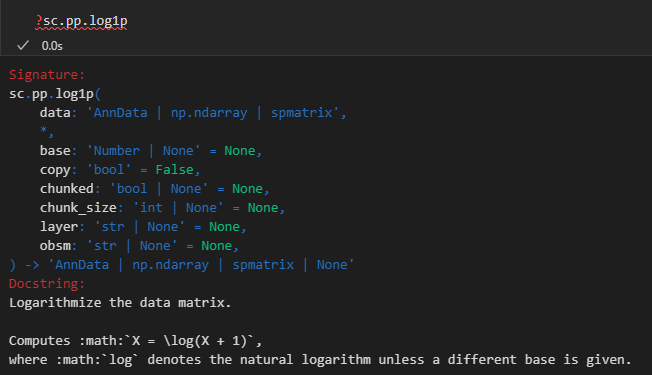
紧接着就是计算高变gene
在单细胞数据中,不同细胞类型之间的基因表达差异可能是高度变异基因引起的。识别这些基因对于后续的聚类和差异分析非常重要。
# 计算高变基因
# 识别在不同细胞中具有高度变异的基因,这些基因在细胞间表达差异显著,是进一步分析的重点
sc.pp.highly_variable_genes(adata, min_mean=0.0125, max_mean=3, min_disp=0.5)
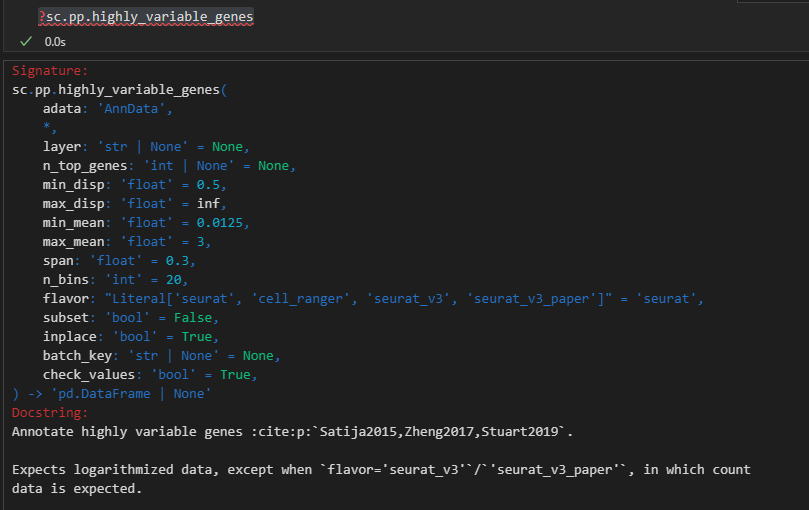
可以数据是取决于n_top_genes的,如果前者没有预设一个值的话,那么过滤筛选的条件就是看min、max那些指标了;
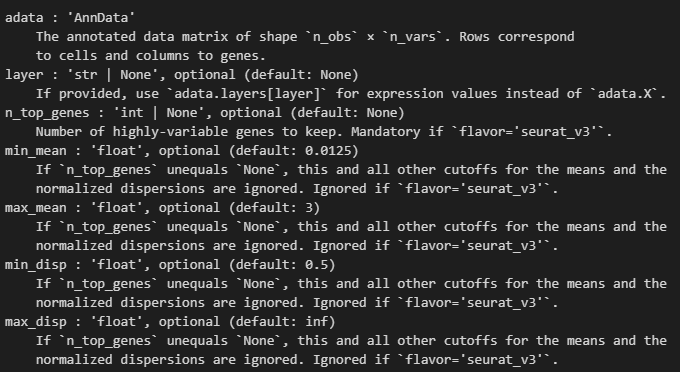
可以看到使用的都是一些默认的值
可以看到在执行之后会在adata的var数据框中增加一些column列,最主要的是在var中增加了highly_variable,也就是使用bool值标注了一些是否是高度可变的gene的mark,然后可以用于后续下游分析的时候筛选出来这些gene
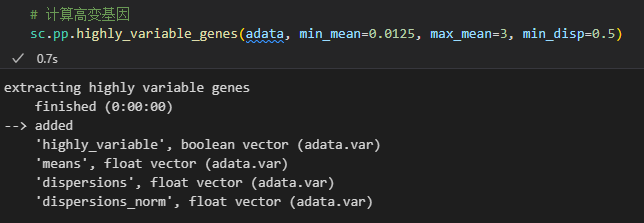
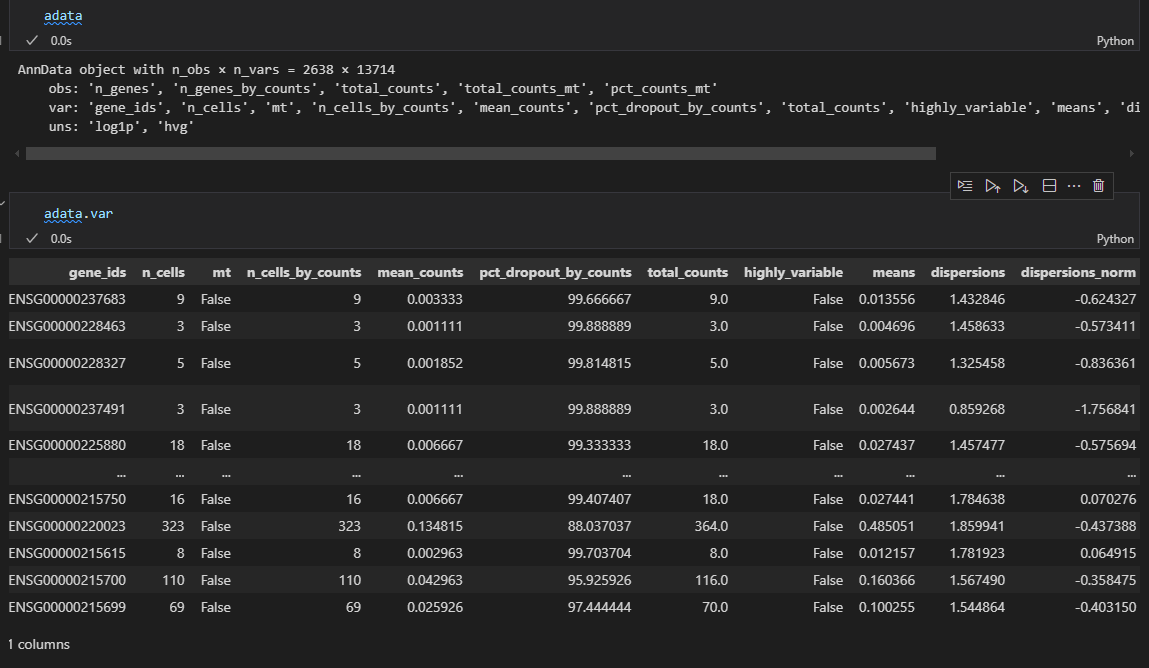
可以看到这里有一列bool列
然后就是对这些gene的可视化(前面找出了高度可变的gene,那就有这些gene与非高度可变gene的一些指标的比较)
因为可视化的是var数据框对象里的column,而var是对gene,即for a gene(在所有cell中)
# 可视化基因离散度与平均表达量之间的关系
sc.pl.highly_variable_genes(adata)
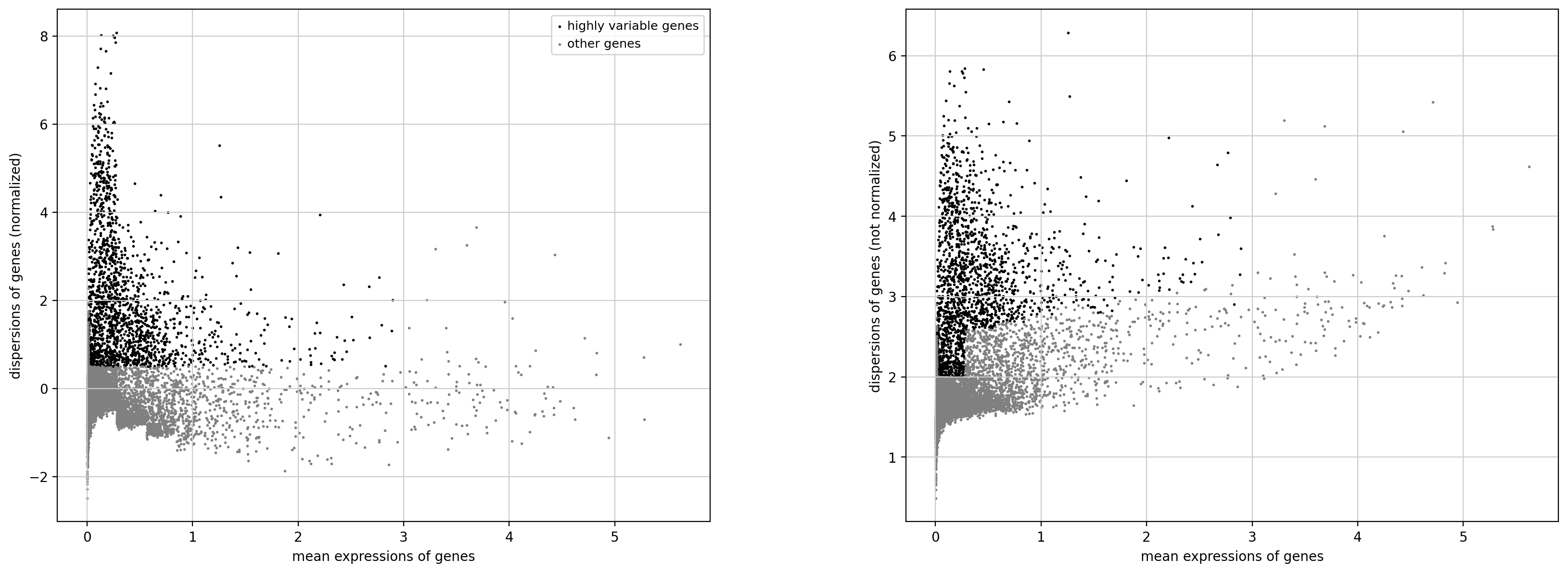
绘制的是(散度,或者是归一化之后的方差)与均值的关系,对于gene
所以数据应该就是对于每一个cell,然后使用的就是所有gene的数据,在对所有的gene进行一个归一化之后的,图上的每一个小点就是一个cell,然后绘制是这个cell中的gene之间的var vs mean
注意在运行过程中可以时刻保留一个备份接口,其实就是相当于R中的数据备份
# 利用view操作对AnnData做一个备份:
adata.raw = adata
# 这样就能保证你在下游操作失误或想回溯原始数据时具有方便的接口
adata.raw
然后就是对于这些高变gene的一些处理:保留高变gene
# 过滤,仅保留高变基因,获取只有特异性基因的数据集
adata = adata[:, adata.var.highly_variable]
# 回归排除细胞counts总数与线粒体含量对下游分析造成的影响
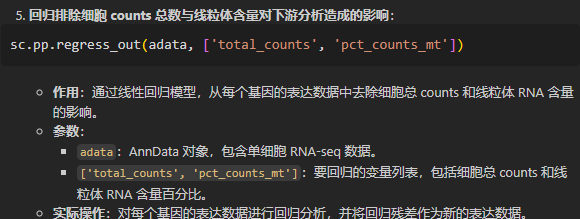
sc.pp.regress_out(adata, ['total_counts', 'pct_counts_mt'])
# 计算每个基因的单位方差,剪辑标准偏差超过10的值,完成“缩放”过程,
# 让每个基因在下游参与降维分群过程中处于“平等”的地位。
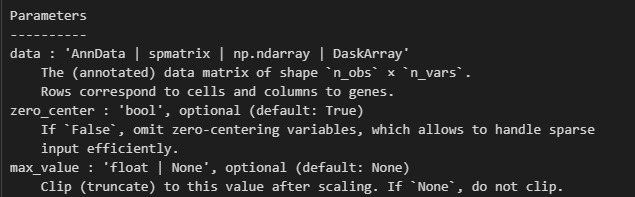
sc.pp.scale(adata, max_value=10)
官网教程中的描述是:
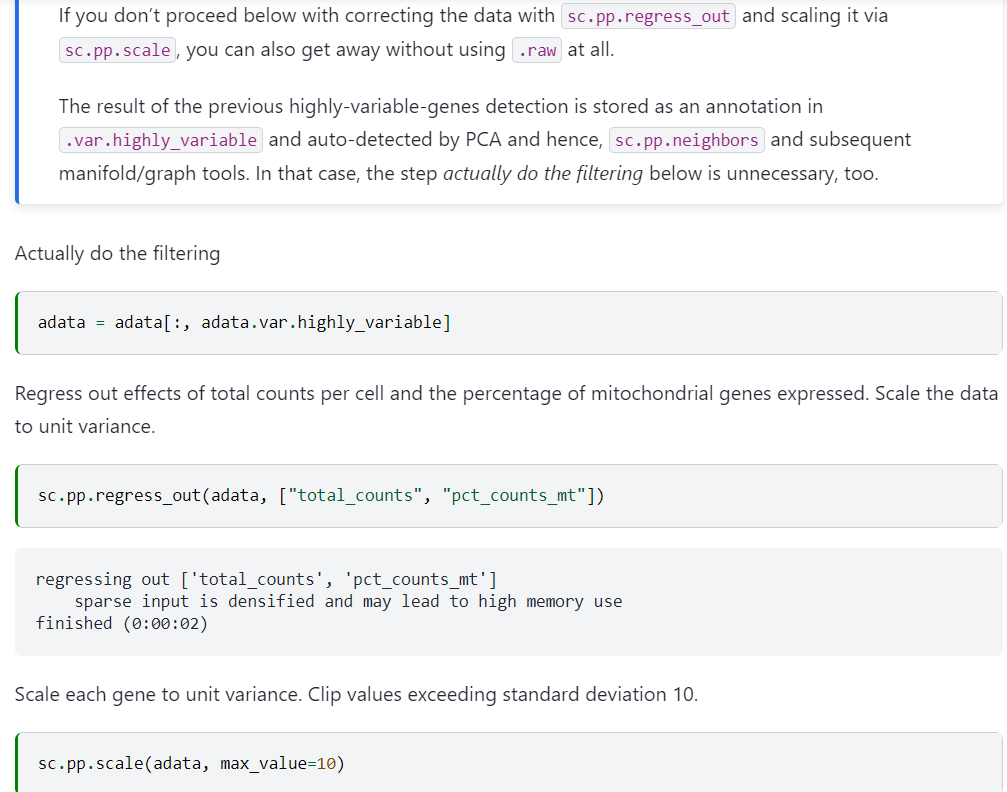
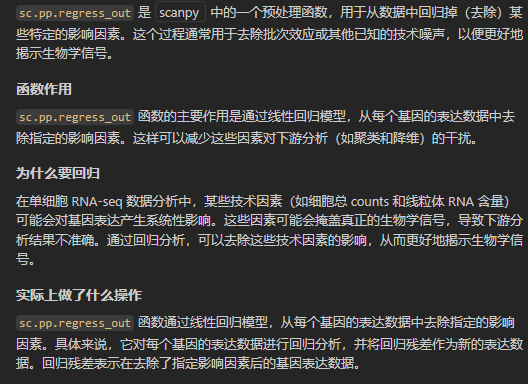
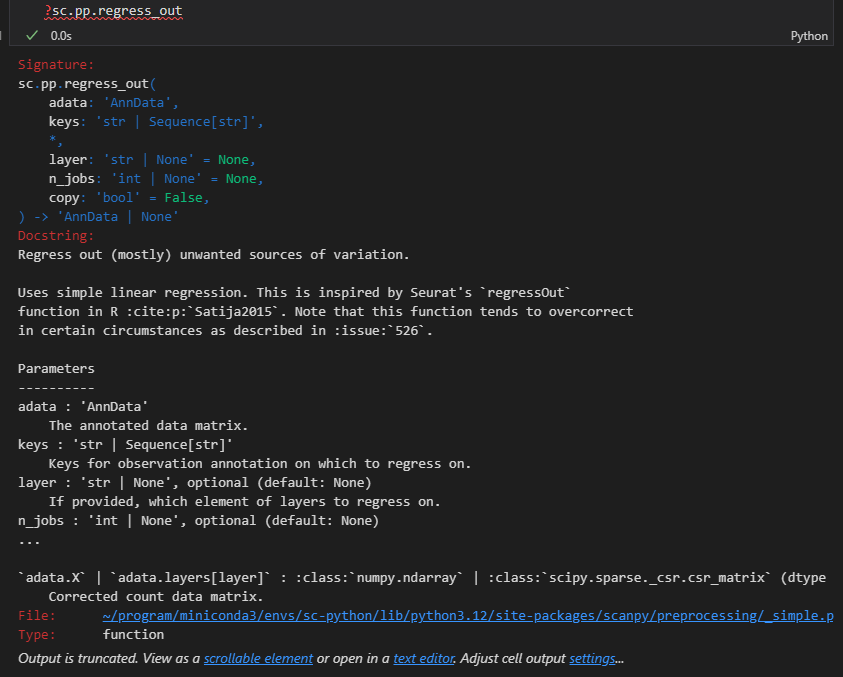
事实上我们可以看到这里需要回归排除的数据都是obs轴上的数据,也就是for a cell的数据
scale函数就很简单:就是均值为0、方差为1的归一化(标准化)
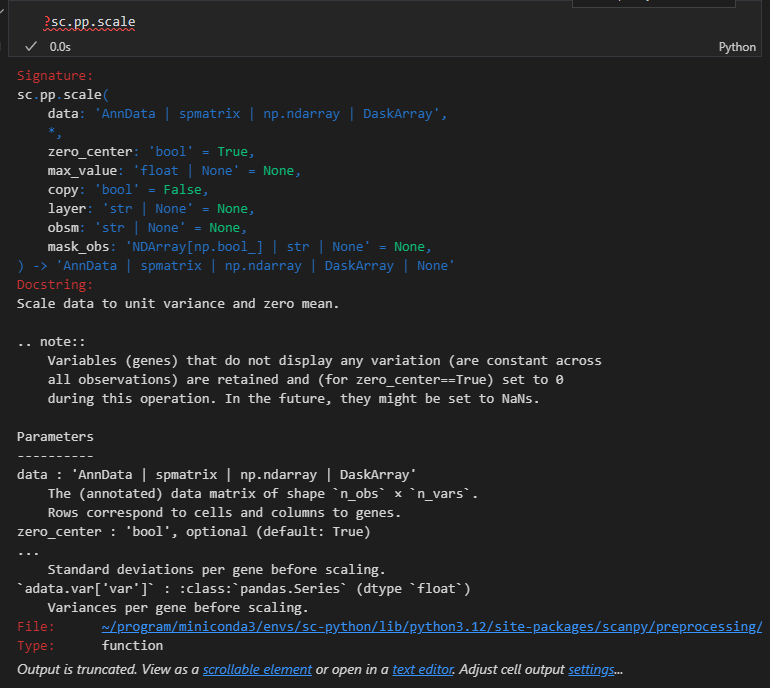
总体就是



然后就可以进行降维分析了,首先是PCA
# 完成主成分分析(principal component analysis, PCA)
sc.tl.pca(adata, svd_solver='arpack')
# 可视化:
sc.pl.pca(adata, color='CST3')
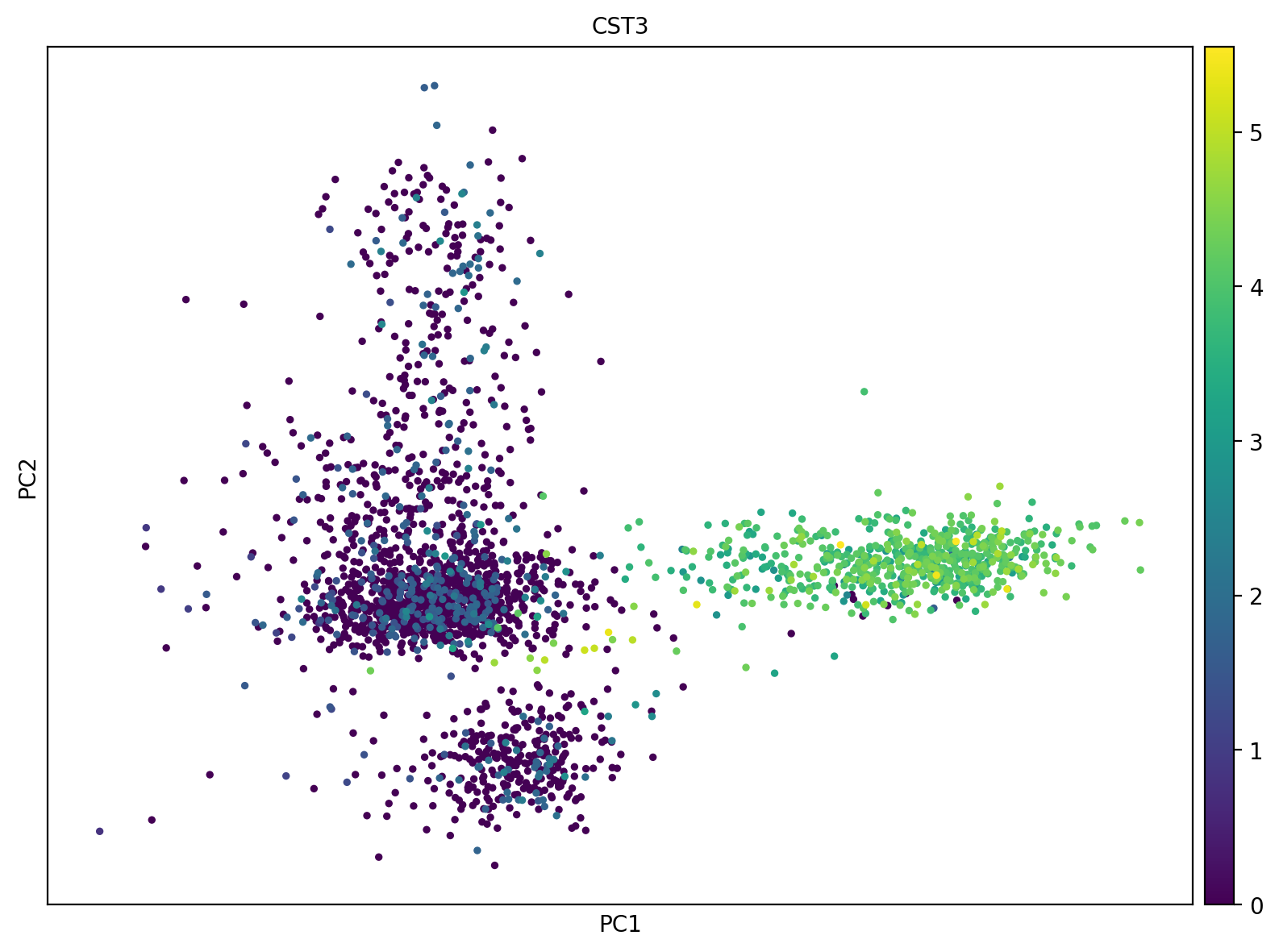
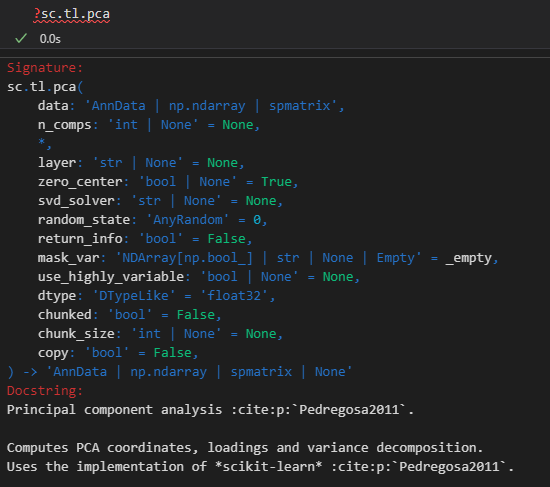
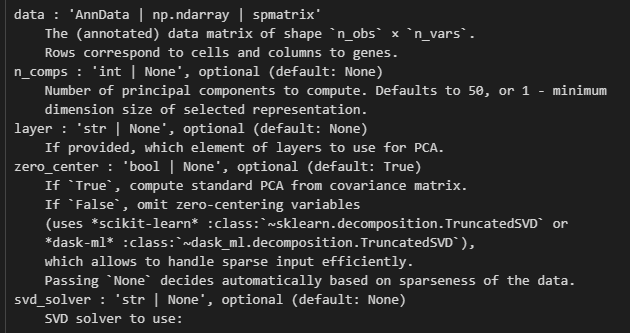
计算的主成分默认至50个,
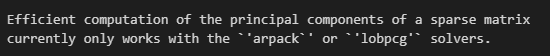
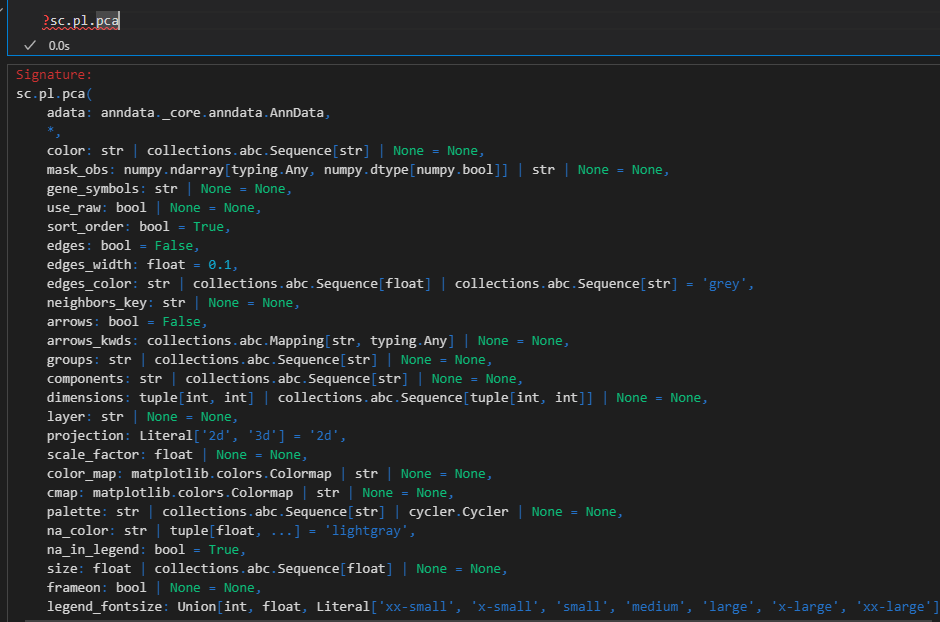

color可用于obs的cell或var的gene
然后在做了PCA之后,就是查看各个主成分对于方差的贡献度
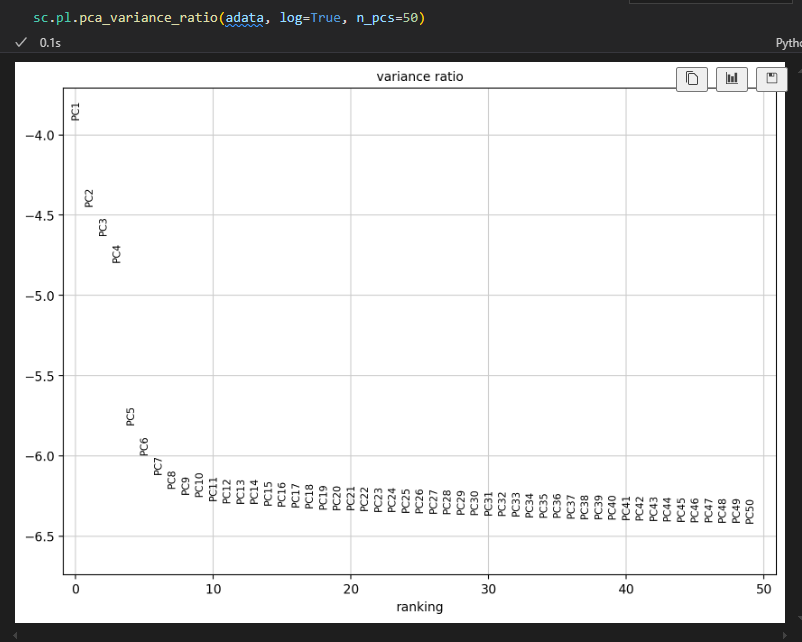
# 计算每个PC对于数据方差的贡献度,决定了下游用于降维、聚类的PC数量:
# 类似于Seurat中的ElbowPlot
sc.pl.pca_variance_ratio(adata, log=True)
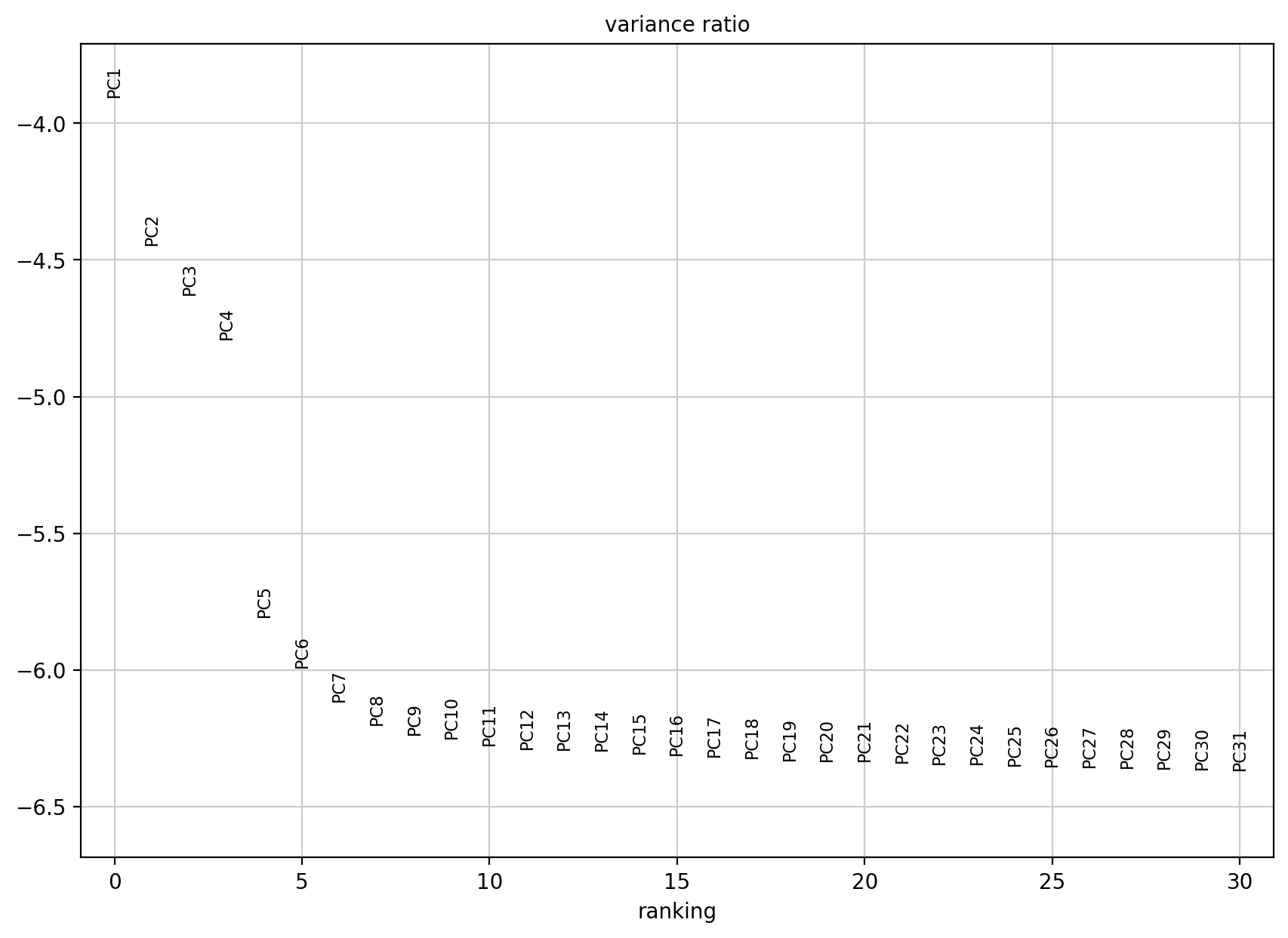
如果不是在log范围内展示数据的话,就是1单纯查看原始的数值,而不是log之后的负值
sc.pl.pca_variance_ratio(adata, log=False)
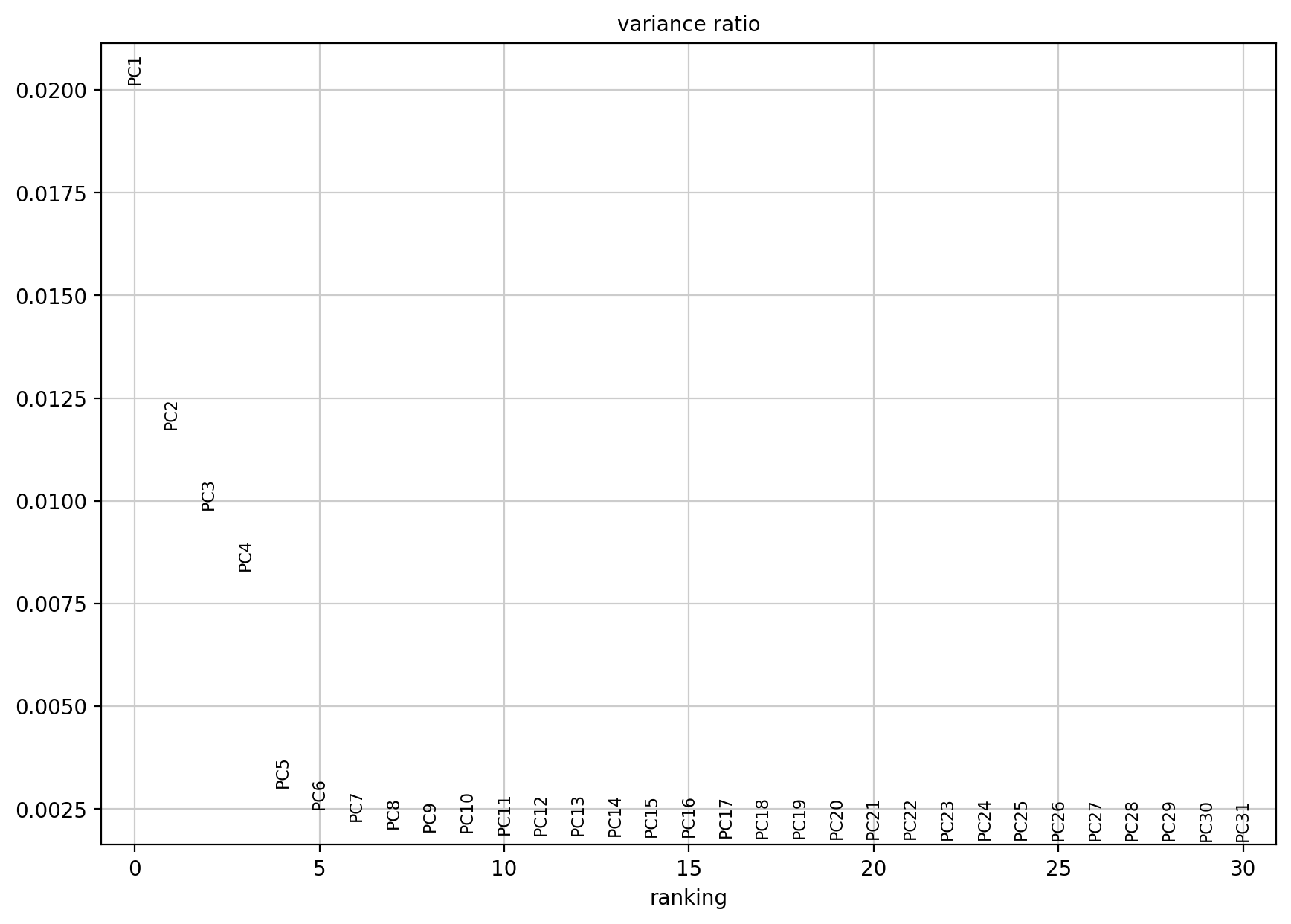
当然在实际绘制的时候是可以绘制多达50个主成分PC的
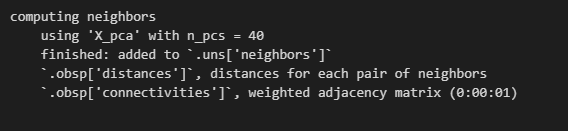
# 利用PCA代表矩阵计算neighborhood graph,
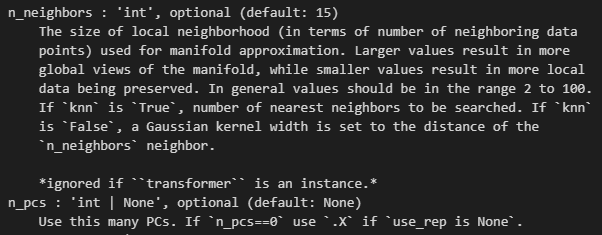
# n_neighbors可以根据上图中变平缓的转折点选取,一般在10~20之间:
sc.pp.neighbors(adata, n_neighbors=10, n_pcs=40)
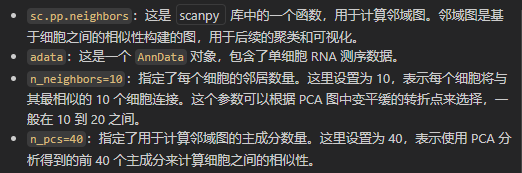
前面使用PCA,所以我们有PCA的tl再pl可视化,当然我们也可以使用umap或tsne
总之就是少用PCA
# 在两种降维方法种,作者更倾向于umap:
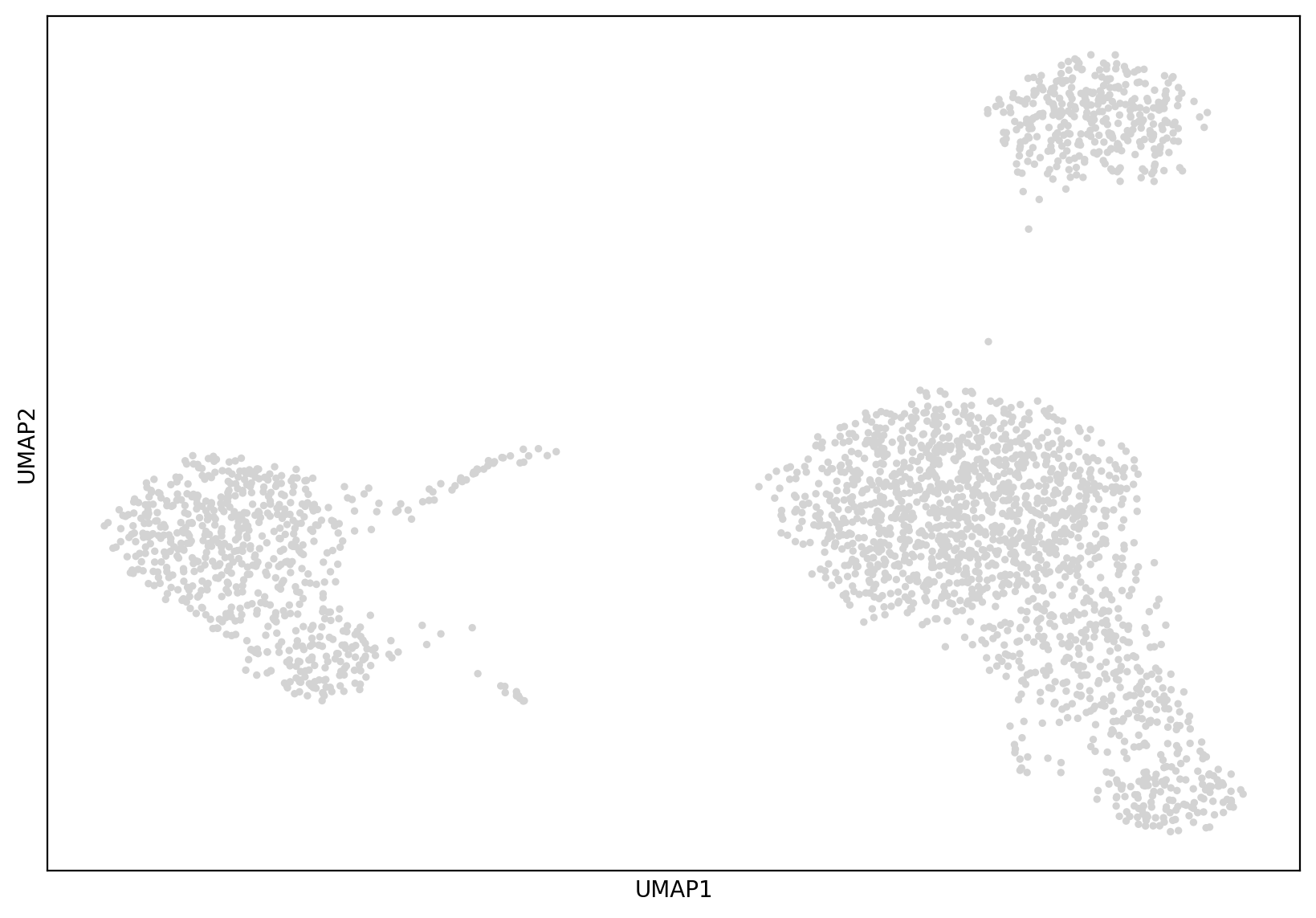
sc.tl.umap(adata)
# 查看降维结果
sc.pl.umap(adata)
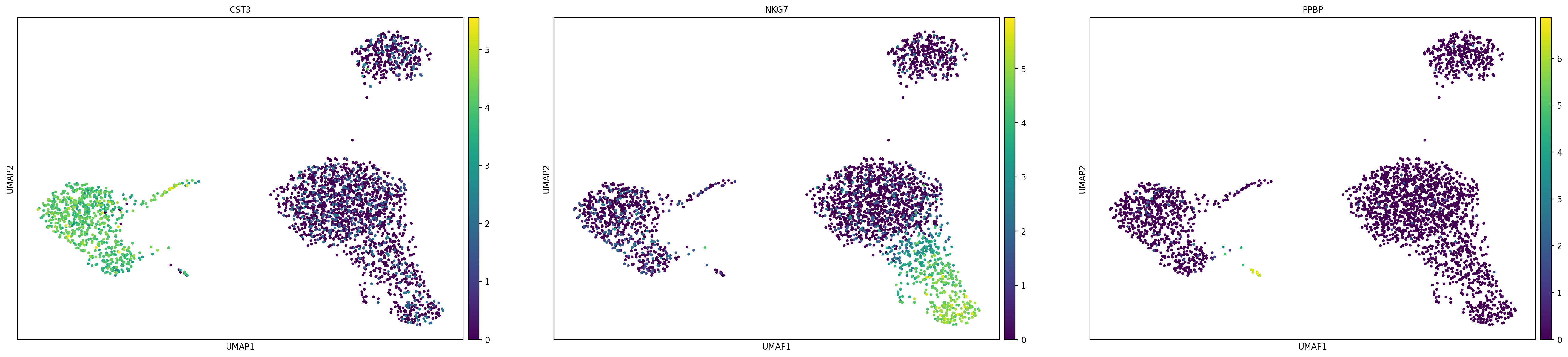
# 可以直接在umap的结果上绘制基因表达量,类似于Seurat中的FeaturePlot
sc.pl.umap(adata, color=['CST3', 'NKG7', 'PPBP'])
# 当你想回溯上一个代码框的原始数据版时:
sc.pl.umap(adata, color=['CST3', 'NKG7', 'PPBP'], use_raw=False)

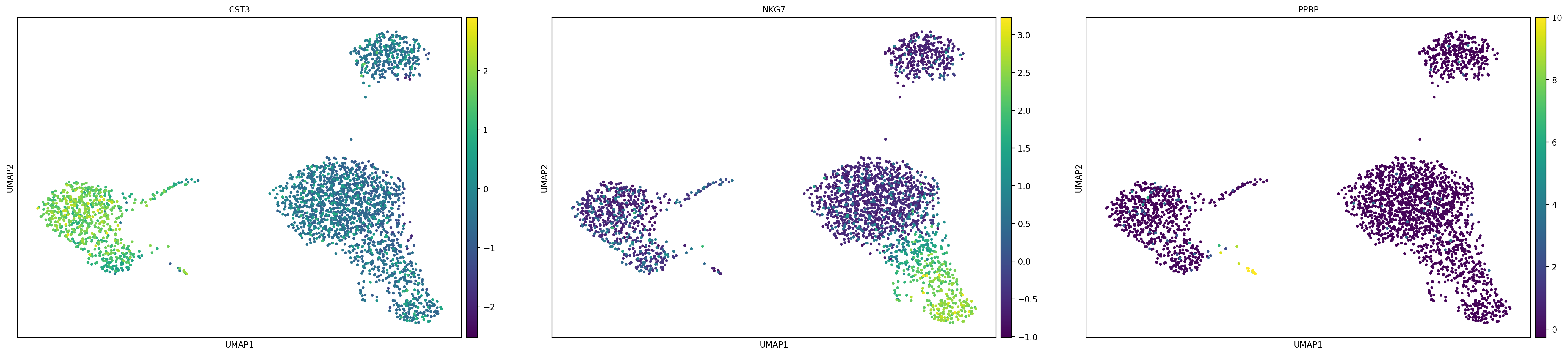
其实我们可以看到仅仅只是数值上的差异而已,就是前面var对于gene的表达值的一系列操作
着色程度上也只是相对于数值而言的

目前还没有计算出各个细胞类群,下面进行聚类
然后根据提示
/tmp/ipykernel_986980/2386482808.py:3: FutureWarning: In the future, the default backend for leiden will be igraph instead of leidenalg.
To achieve the future defaults please pass: flavor="igraph" and n_iterations=2. directed must also be False to work with igraph's implementation.
sc.tl.leiden(adata)
提示之中需要修改的地方:

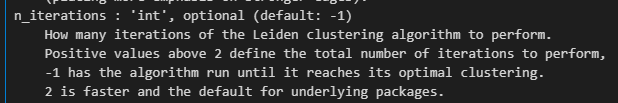
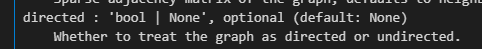
sc.tl.leiden(adata,flavor="igraph",n_iterations=2,directed=False)
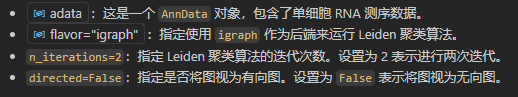
计算出来的结果是不一致的:采用后者
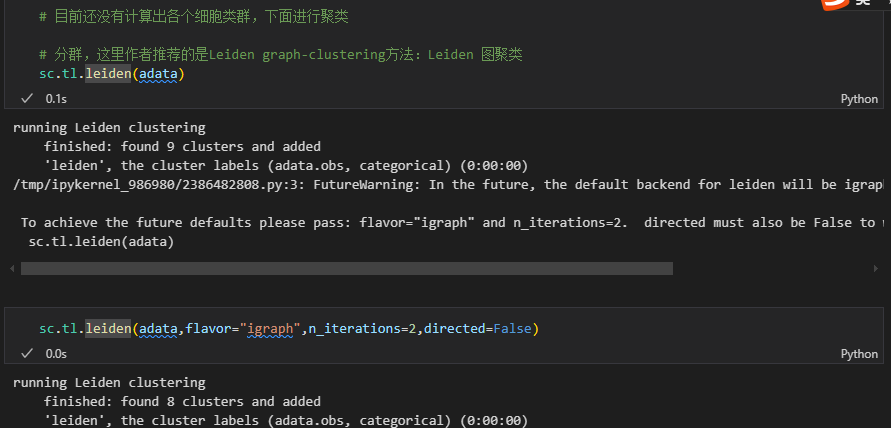
根据上面的结果我们可以看到分出了8个亚群
然后其实在obs的结果列中也展示出来了:
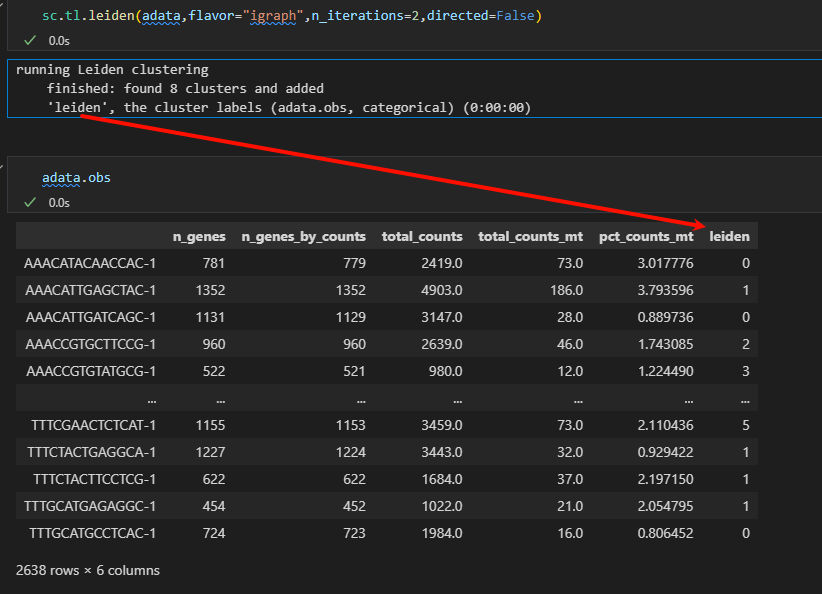
# 一共分为了八个亚群
sc.pl.umap(adata, color=['leiden'])
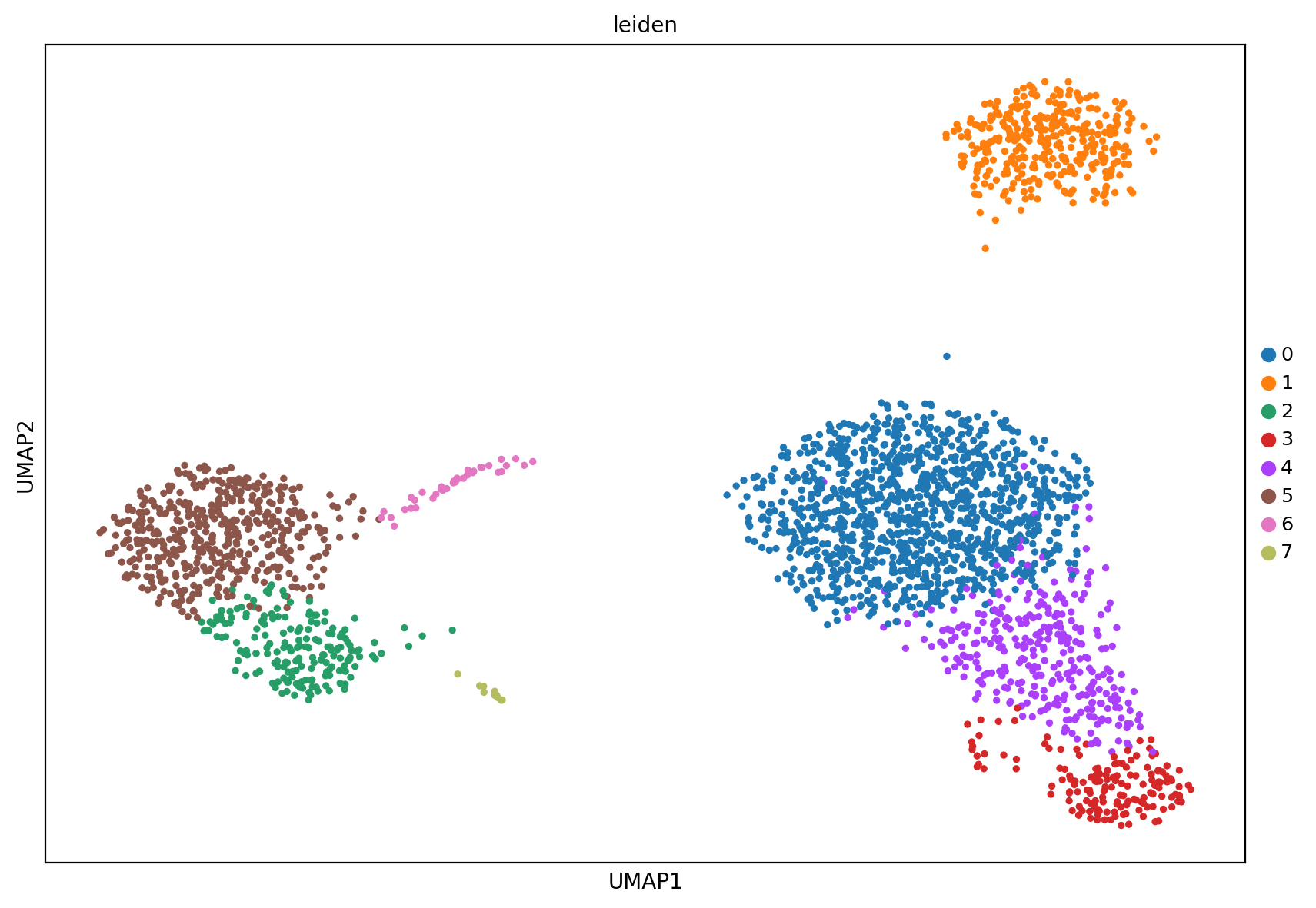
所以这里只是使用了obs的某一列来展示,也就是前面注释的结果列
然后紧接着就是检索生物学标记gene:
先计算每个 leiden 分群中高度差异基因的排名,取排名前 25 的基因。默认情况下,使用 AnnData 的 .raw 属性。
最简单和最快的方法是 t 检验。
# 检索生物学标记基因
# 先计算每个 leiden 分群中高度差异基因的排名,取排名前 25 的基因。默认情况下,使用 AnnData 的 .raw 属性。
# 为marker基因的计算做ranking,最快的是t检验:最简单和最快的方法是 t 检验
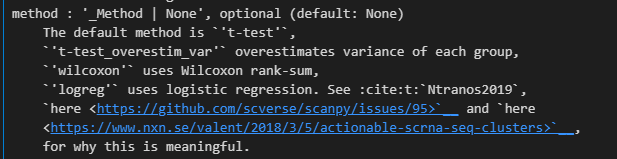
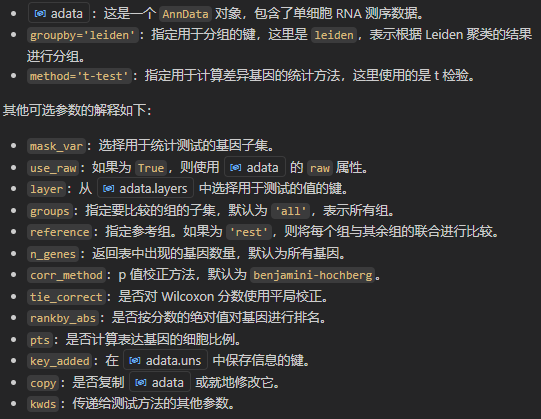
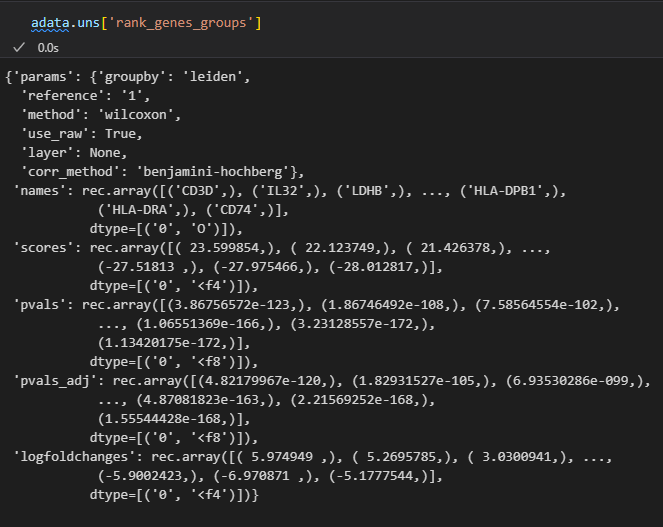
sc.tl.rank_genes_groups(adata, 'leiden', method='t-test')
然后结果添加到了uns结构slot中
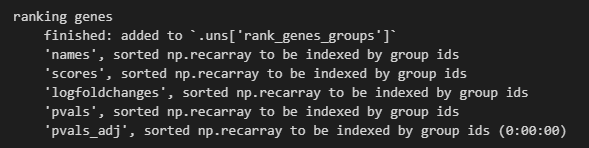
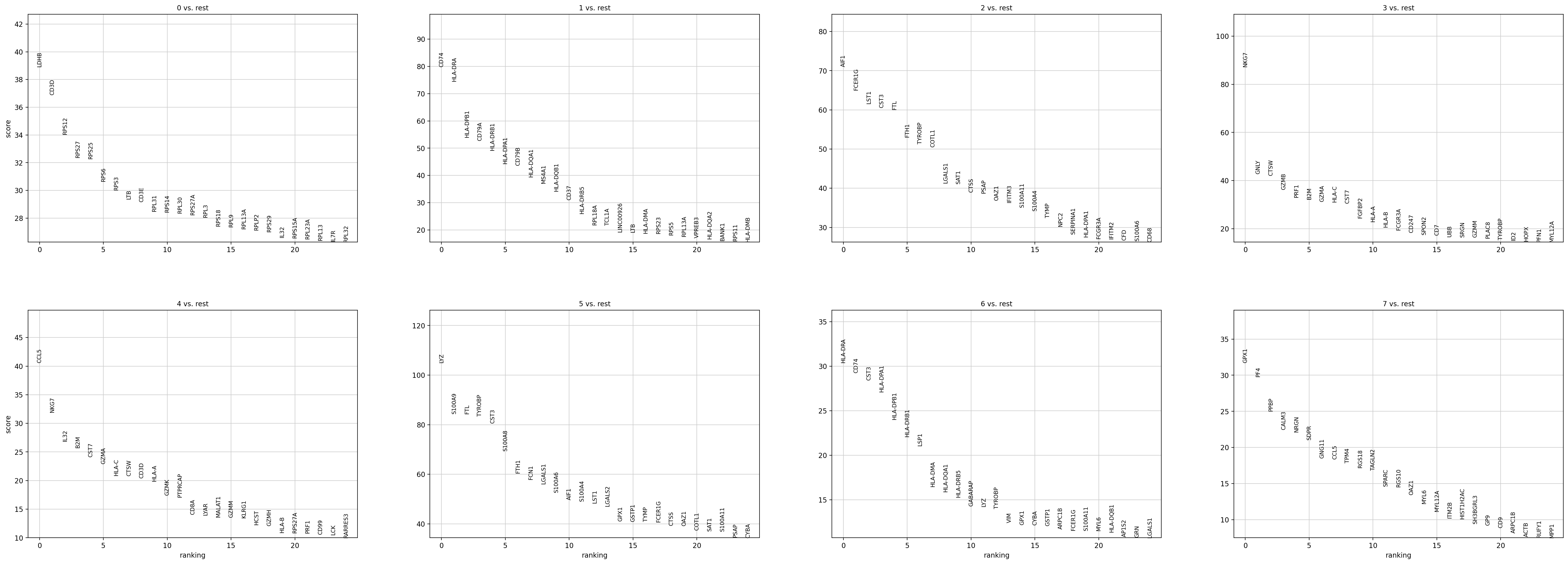
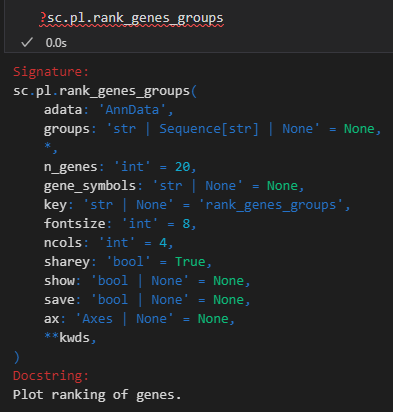
sc.pl.rank_genes_groups(adata, n_genes=25, sharey=False)

# Wilcoxon rank-sum 是作者最推荐的方式,此外还有MAST, limma, DESeq2, diffxpy, logreg等方式
sc.tl.rank_genes_groups(adata, 'leiden', method='wilcoxon')
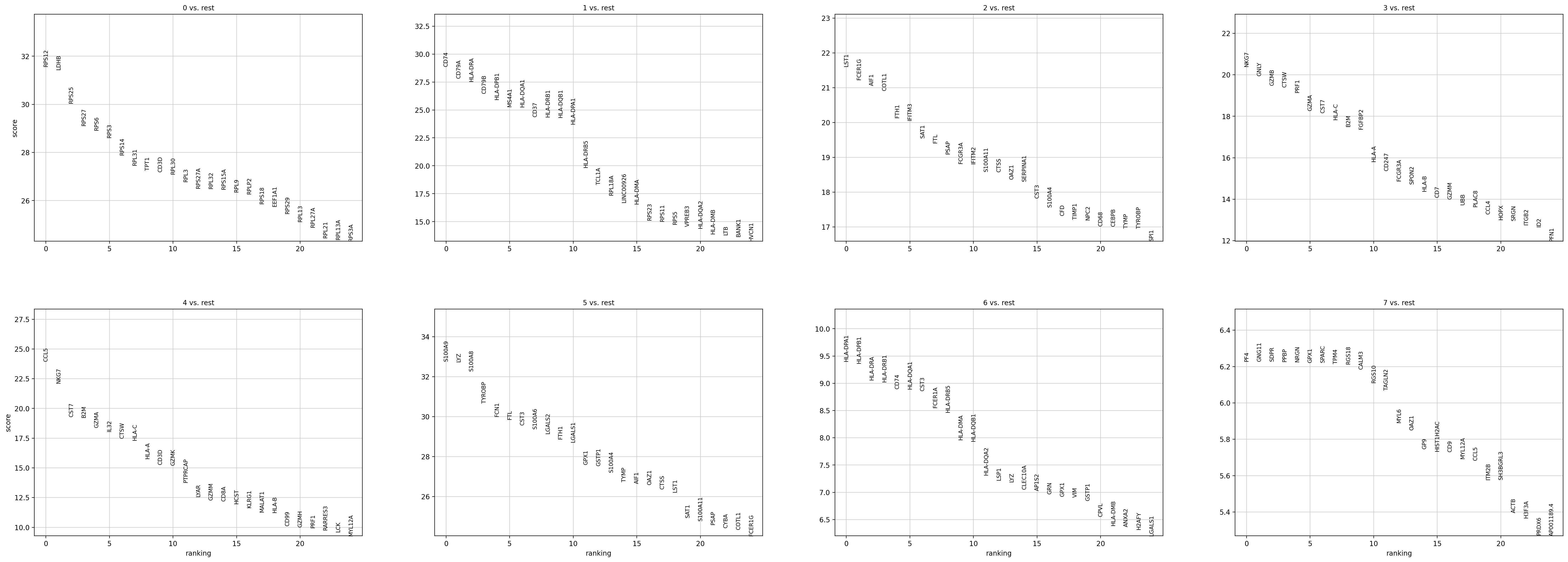
sc.pl.rank_genes_groups(adata, n_genes=25, sharey=False)
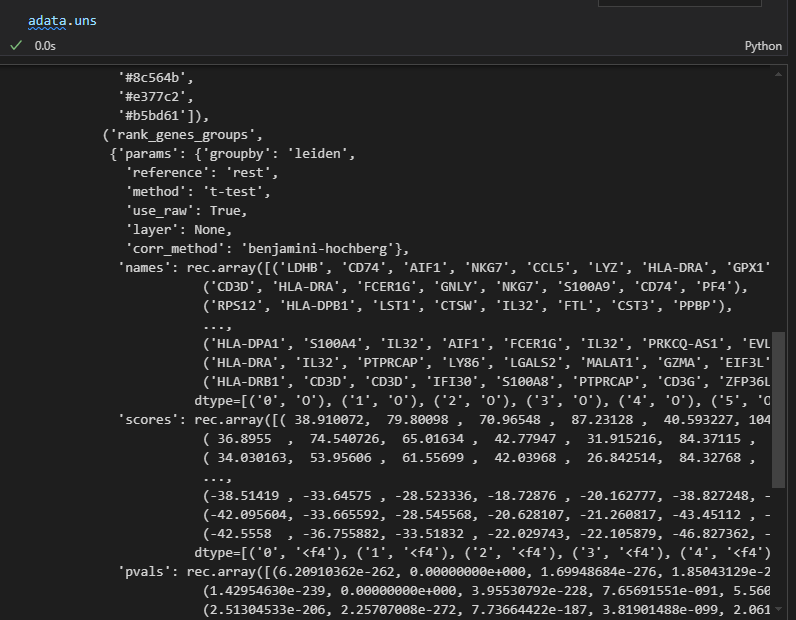
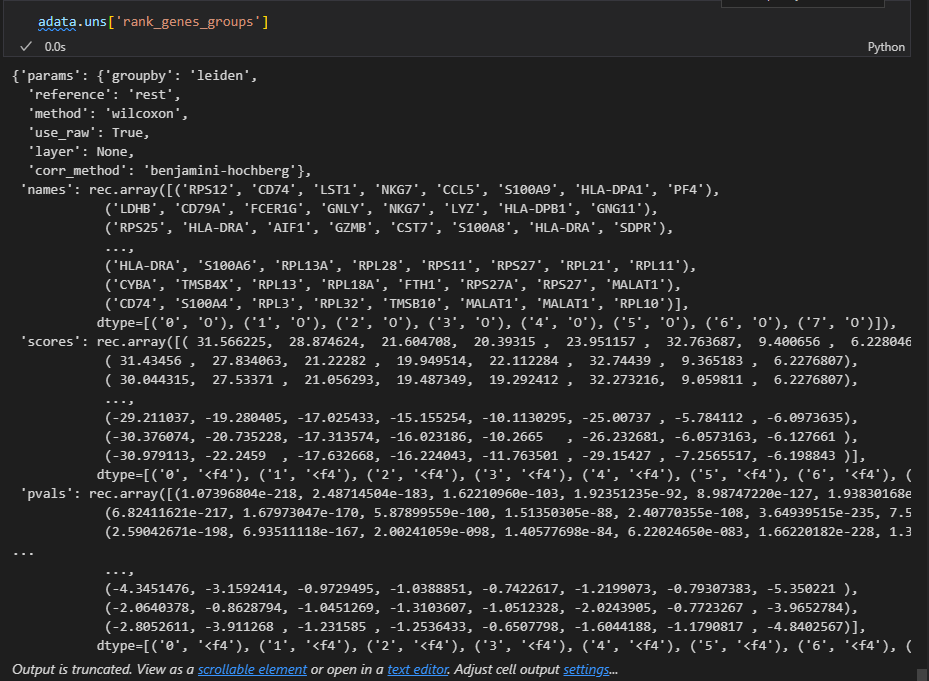
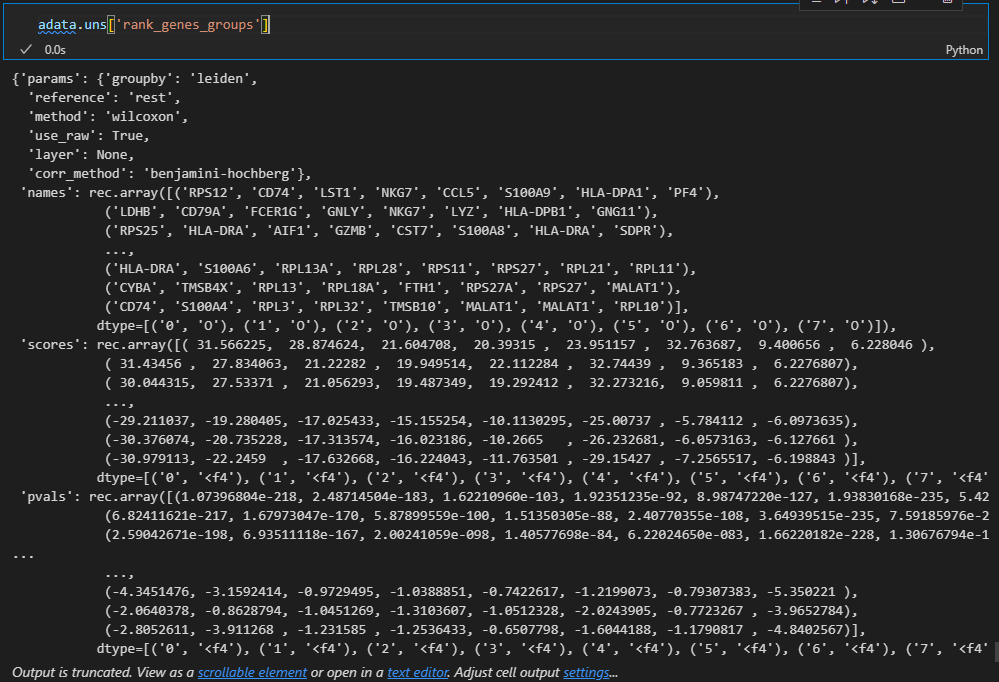
因为结果就存储在uns slot中,所以我们可以到slot这个数据框中进行查看

准确的说是下面的这个复合数据:
然后里面的每一个属性都是一个数据框,比如说是marker gene
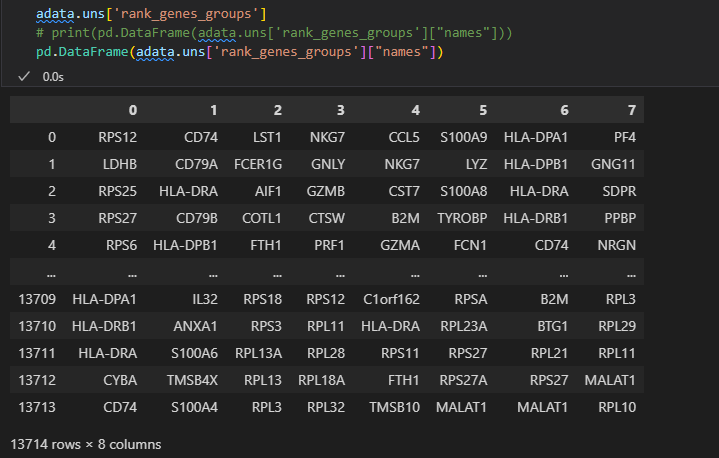
# 这是刚才计算出的每个cluster的top5的marker gene
pd.DataFrame(adata.uns['rank_genes_groups']['names']).head(5)
前面是找到了8个簇,即0-7
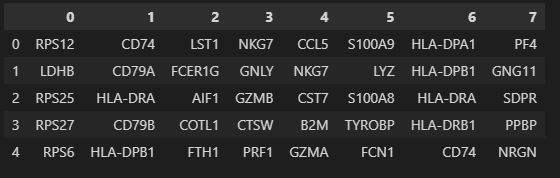
# 查看这些marker在所属cluster中得到的分值:
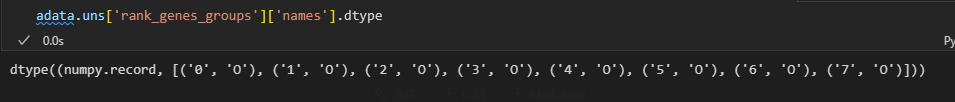
result = adata.uns['rank_genes_groups']
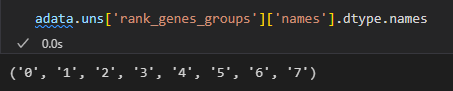
groups = result['names'].dtype.names #分组名称,实际上就是簇名cluster
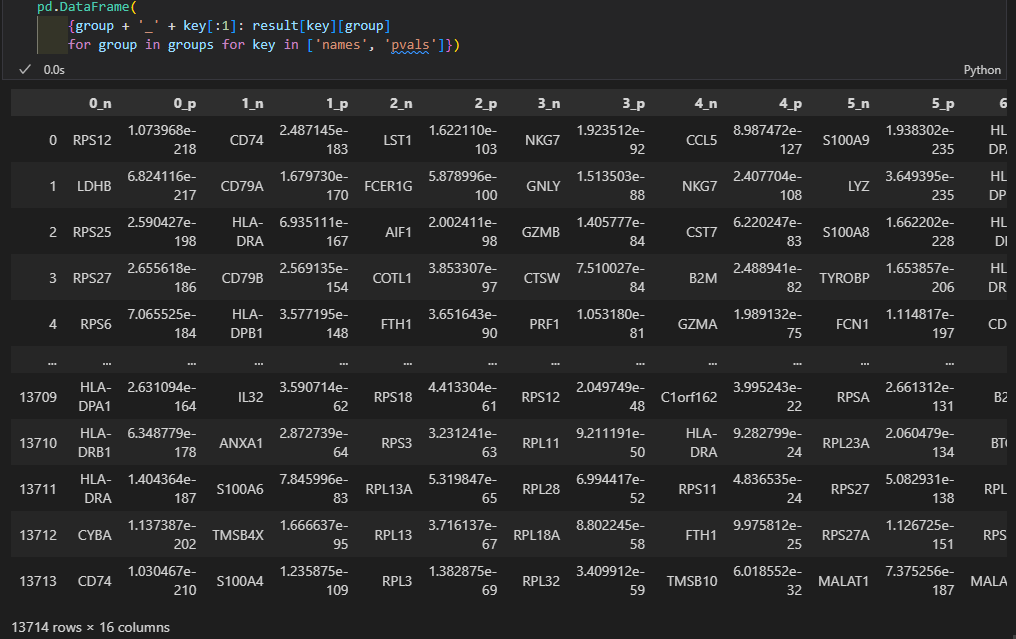
pd.DataFrame(
{group + '_' + key[:1]: result[key][group]
for group in groups for key in ['names', 'pvals']}).head(5)
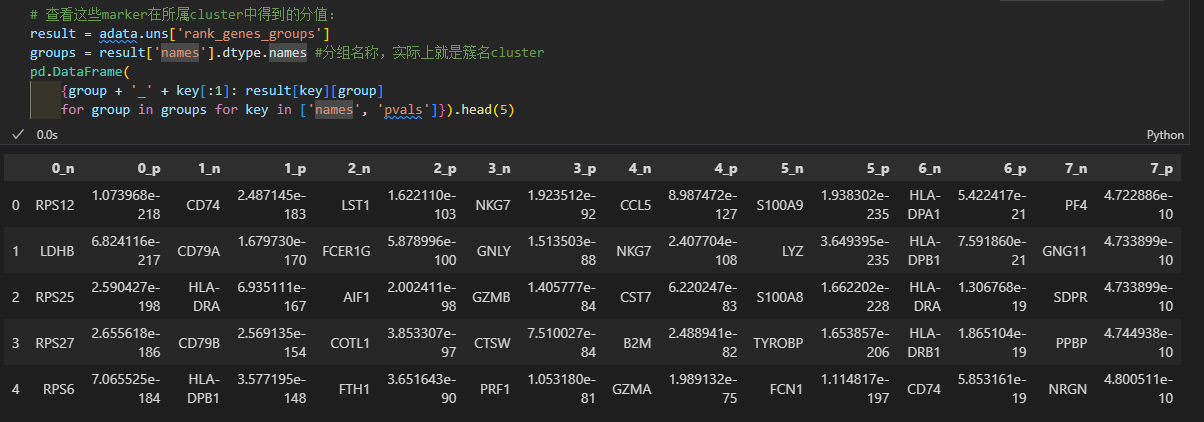
0-7是group,也就是分群名,也就是cluster簇;
然后key[:1]实际上是采用了names以及pvals的第一个字母;
pd里面实际上是一个字典推导式:仔细理解即可
键值对是group + ‘_’ + key[:1]: result[key][group],循环的推导式是for group in groups for key in [‘names’, ‘pvals’]

再然后其中的names键值列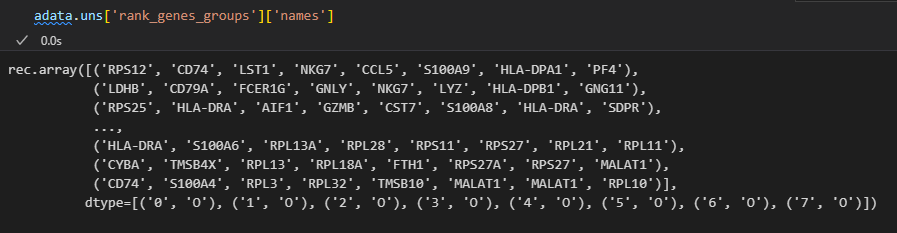
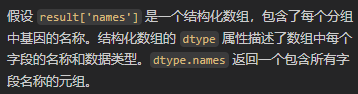
当然每一个分组group/每一个簇展现出的gene有很多,我们可以选择将其可视化
# 画出cluster0的前八个marker基因
sc.pl.rank_genes_groups_violin(adata, groups='0', n_genes=8)
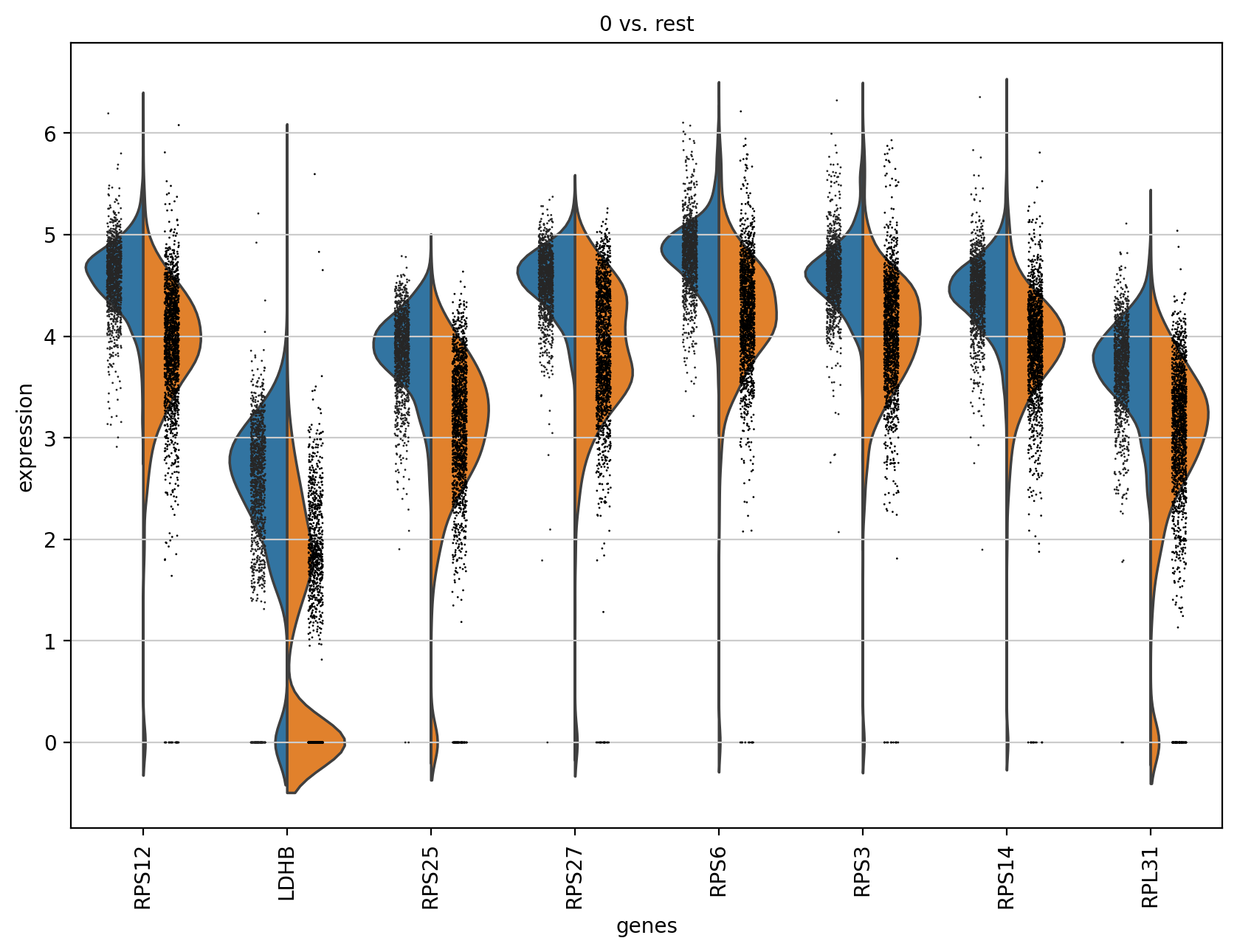
此处是使用了cluster0也就是分组0的marker gene,然后拿这个gene的表达值在cluster0以及其他cluster分组之间进行比较,因为是cluster0的marker,所以我们可以看到cluster0也就是左边的表达值比其他cluster的表达值分布还高

前面的某个分组cluster比较其他分组cluster的情况,可以使用pl直接绘制;
但是如果是两两cluster之间的单独比较,需要先tl计算再pl可视化:
# 单独的两个cluster之间也可以进行marker计算:
sc.tl.rank_genes_groups(adata, 'leiden', groups=['0'], reference='1', method='wilcoxon')
sc.pl.rank_genes_groups(adata, groups=['0'], n_genes=20)
# 计算出的基因可以直接用于小提琴图可视化
sc.pl.rank_genes_groups_violin(adata, groups='0', n_genes=8)
大概是每次计算的时候存储到.uns[‘rank_genes_groups’]里的数据是对应不一致的,
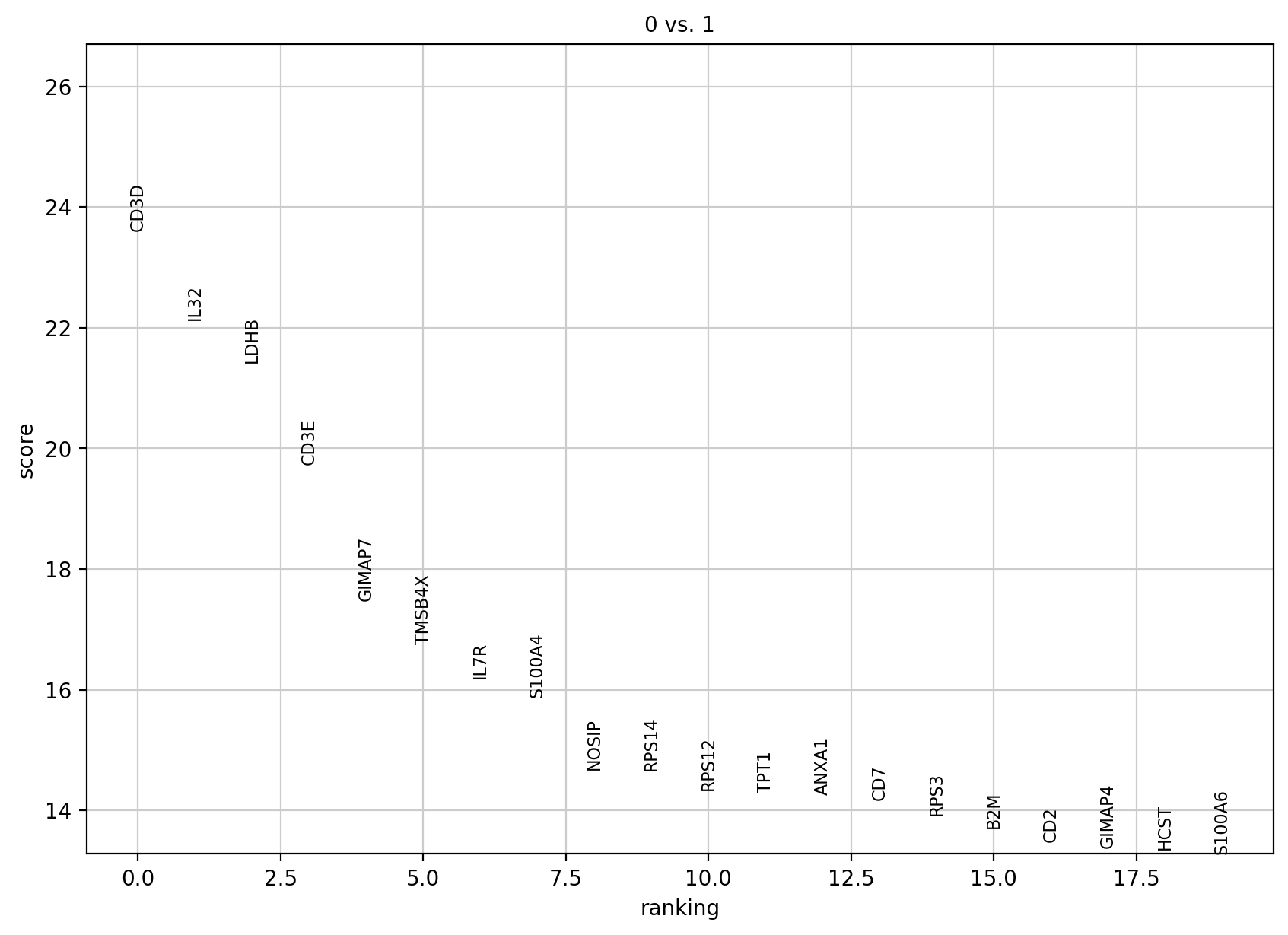
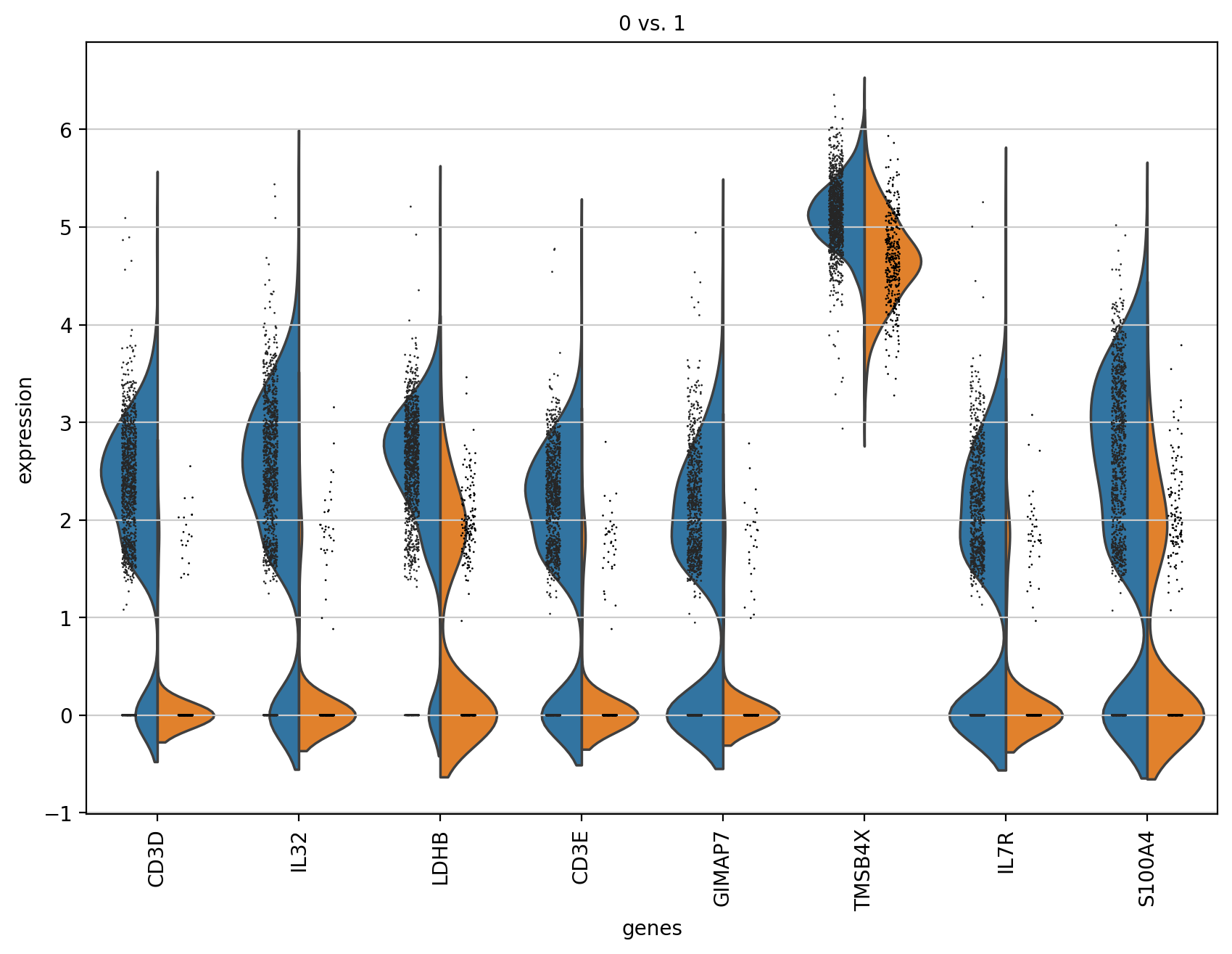
其实可以看到存储的数据发生了变化,所以pl绘制的时候参数没变但是效果是0vs1而不是0vs rest
跨类群比较基因
# 跨类群比较基因
# 画出指定基因在所有cluster中的表达情况:
sc.pl.violin(adata, ['CST3', 'NKG7', 'PPBP'], groupby='leiden')
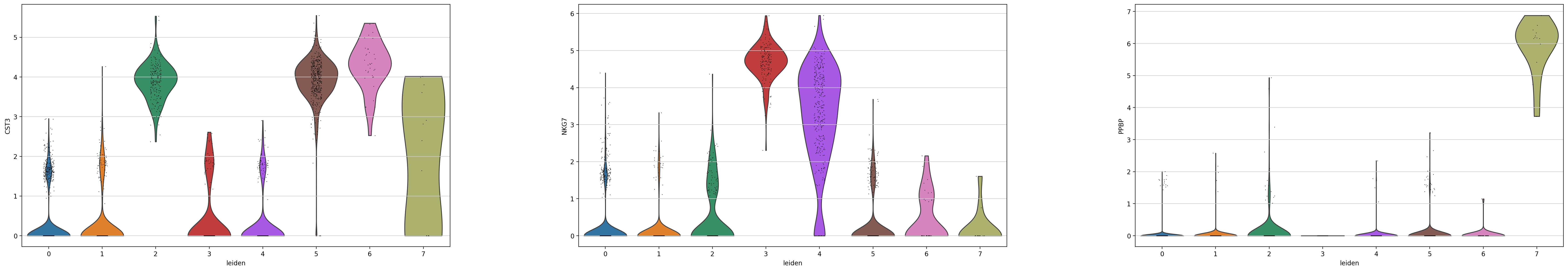
可以查看单独的每个gene在所有的分组cluster中的表达情况的比较
根据已知的细胞标记,注释细胞类型:
如果是自己的数据,需要自己处理注释问题
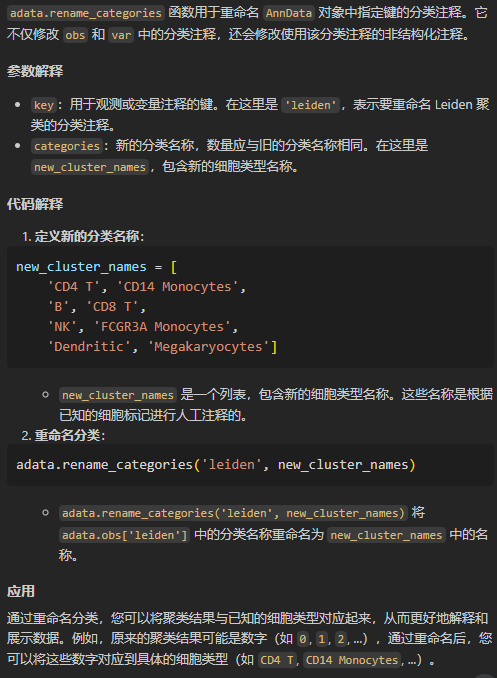
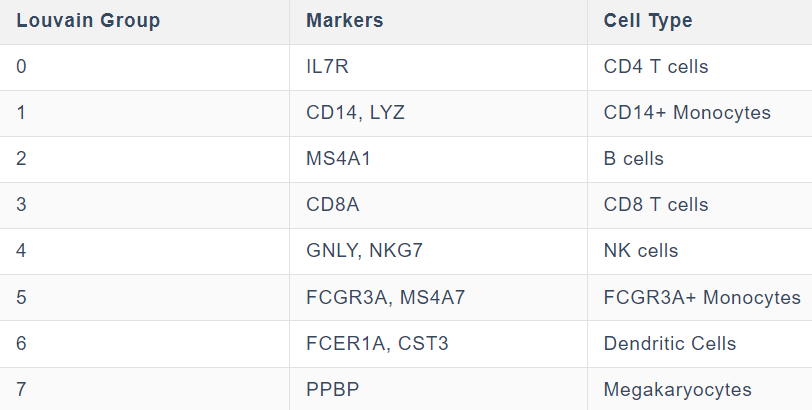
# 根据已知的细胞标记,注释细胞类型(先验,人工注释)
# 重命名各个cluster:
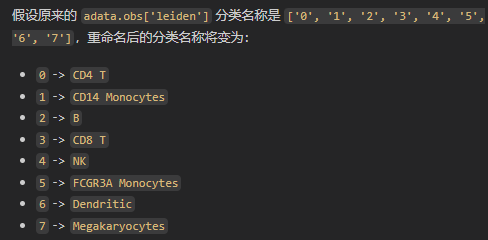
new_cluster_names = [
'CD4 T', 'CD14 Monocytes',
'B', 'CD8 T',
'NK', 'FCGR3A Monocytes',
'Dendritic', 'Megakaryocytes']
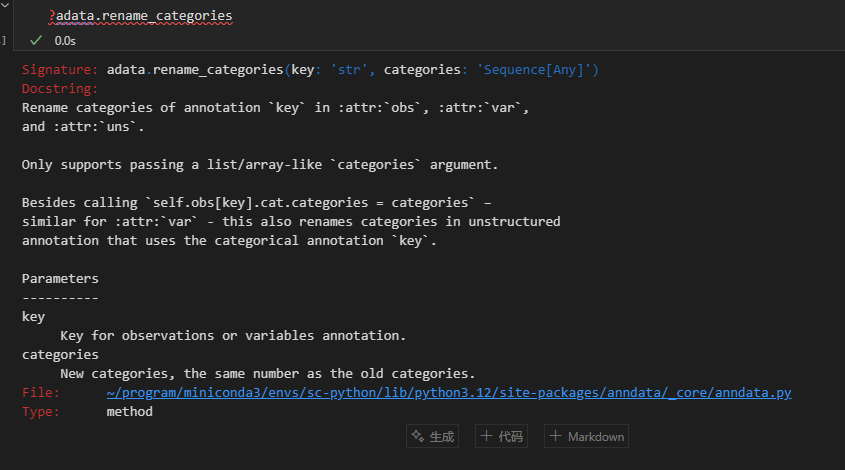
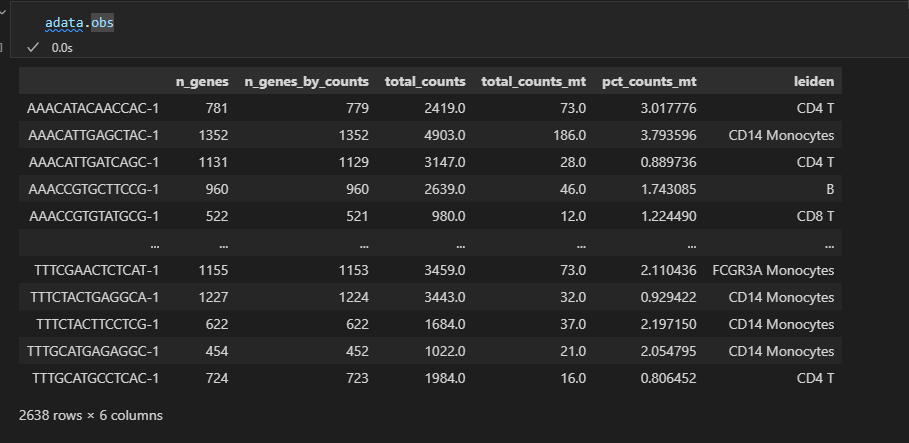
adata.rename_categories('leiden', new_cluster_names)
就是注释leiden列
# 以UMAP的形式进行可视化:
sc.pl.umap(adata, color='leiden', legend_loc='on data', title='', frameon=False, save='.pdf')
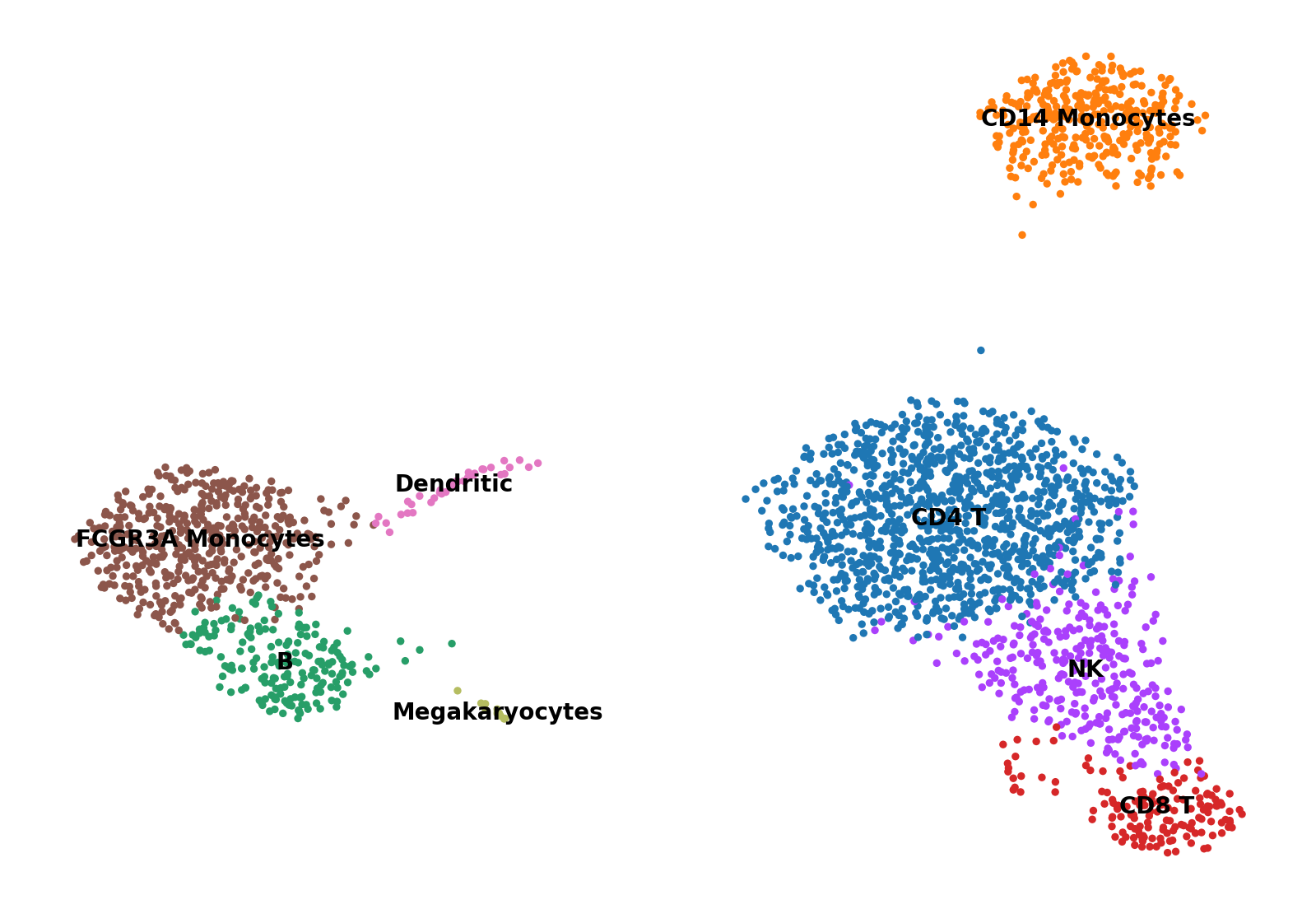
根据已知的标记基因,定义一个标记基因列表供以后参考:
# 作者使用了一些先验的marker,
# 它们是一些常用的细胞标志物:
marker_genes = ['IL7R', 'CD79A', 'MS4A1', 'CD8A', 'CD8B', 'LYZ', 'CD14',
'LGALS3', 'S100A8', 'GNLY', 'NKG7', 'KLRB1',
'FCGR3A', 'MS4A7', 'FCER1A', 'CST3', 'PPBP']
# 以气泡图的形式展示marker基因:
sc.pl.dotplot(adata, marker_genes, groupby='leiden');
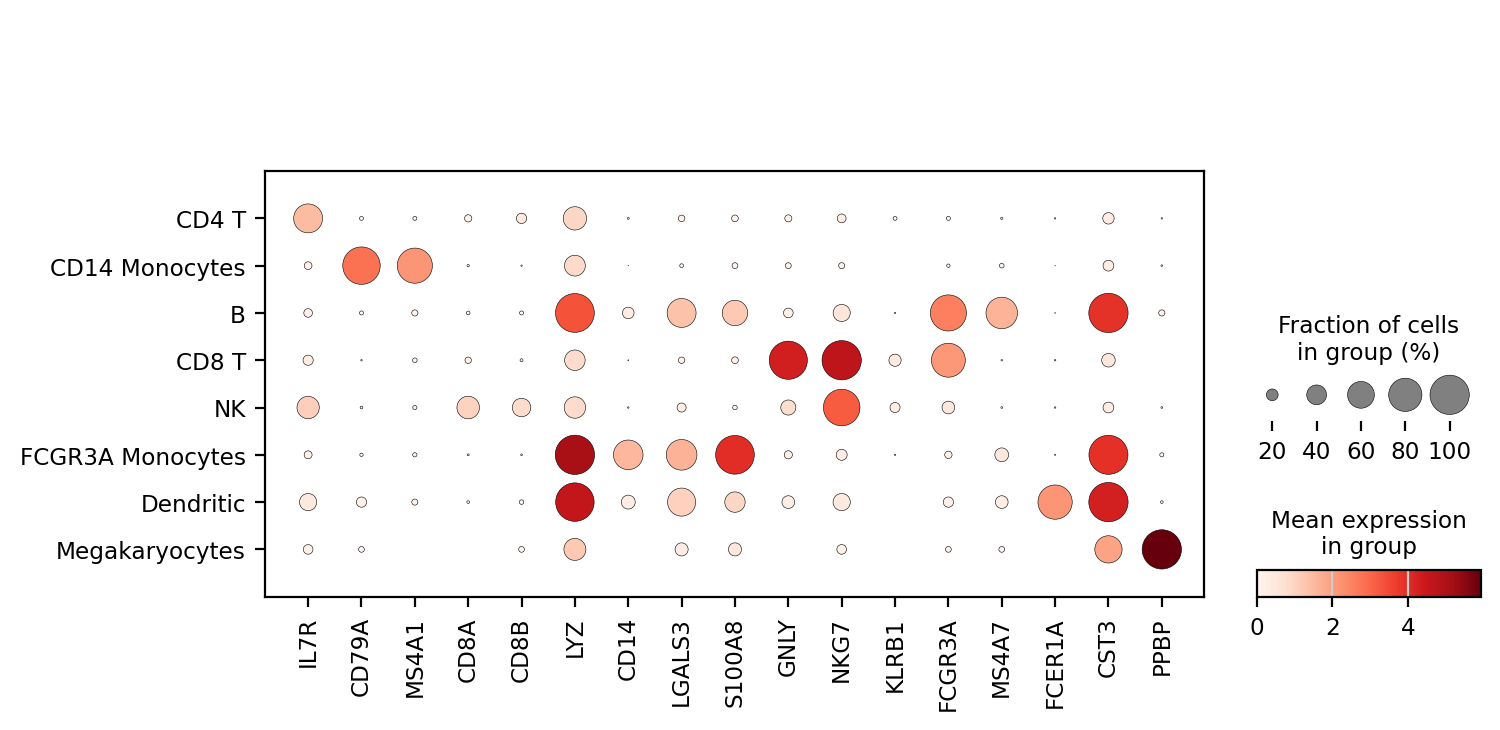


主要是这里的var_names,标注的都是var也就是gene列表中的一个子集

# 以紧凑小提琴的形式展示marker基因:
sc.pl.stacked_violin(adata, marker_genes, groupby='leiden');
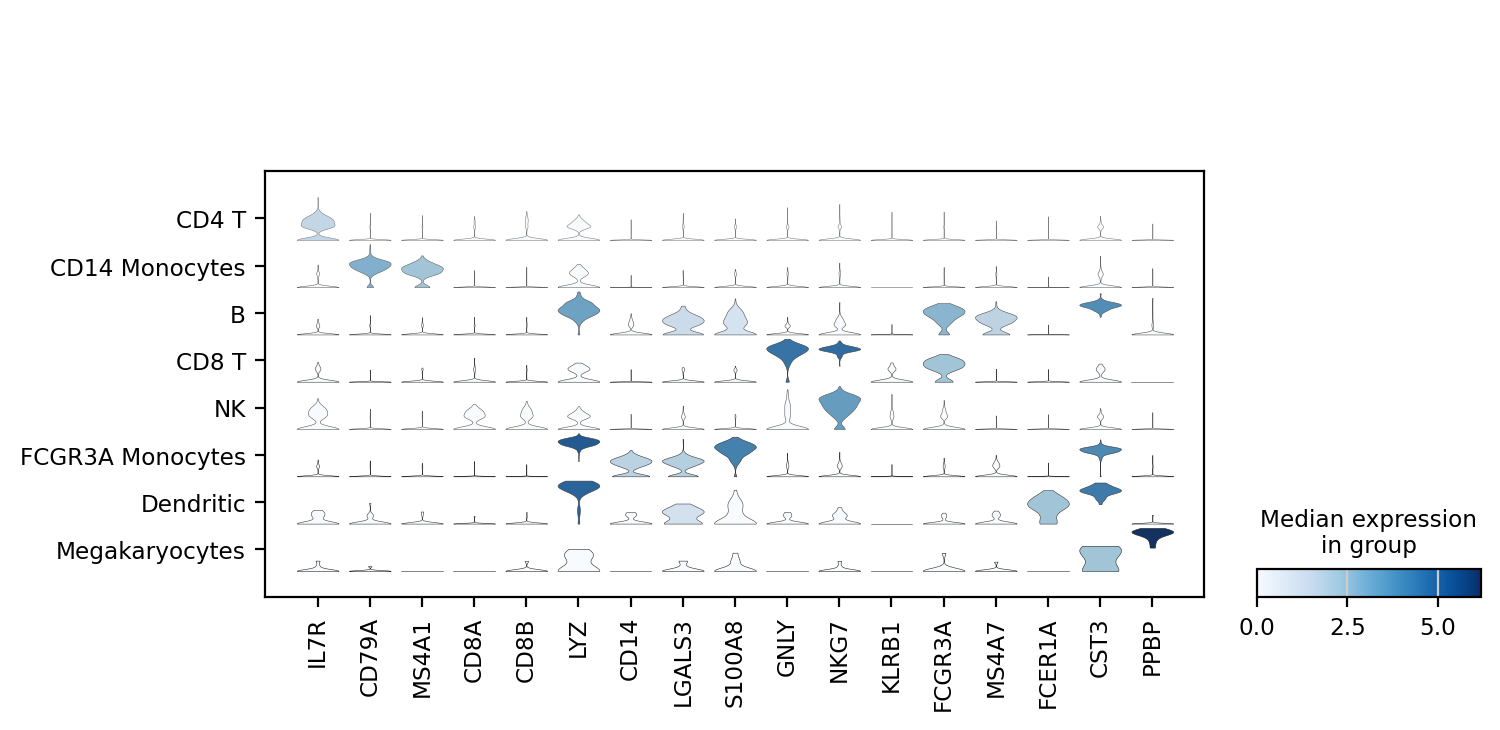
注意这里是没有rotation参数的,有些教程会将这个参数也写进来,但是实际上是错误的
# 保存数据
# 保存压缩文件如果只想将其用于可视化的人共享此文件,减少文件大小的一种简单方法是删除缩放和校正的数据矩阵。
results_file = '/zht/write/pbmc3k.h5ad'
adata.write(results_file, compression='gzip') # 注意,压缩虽然会节省硬盘空间,但是会降低读写速度
# 保存为 h5ad 数据
# 保存原先备份的数据:
adata.raw.to_adata().write('/zht/write/pbmc3k_withoutX.h5ad')
# 读取使用 adata = sc.read_h5ad('./write/pbmc3k_withoutX.h5ad')
# 导出数据子集
# 导出聚类数据
adata.obs[['n_counts', 'louvain_groups']].to_csv('/zht/write/pbmc3k_corrected_louvain_groups.csv')
# 导出PCA数据
adata.obsm.to_df()[['X_pca1', 'X_pca2']].to_csv('/zht/write/pbmc3k_corrected_X_pca.csv')
注意要使用绝对路径

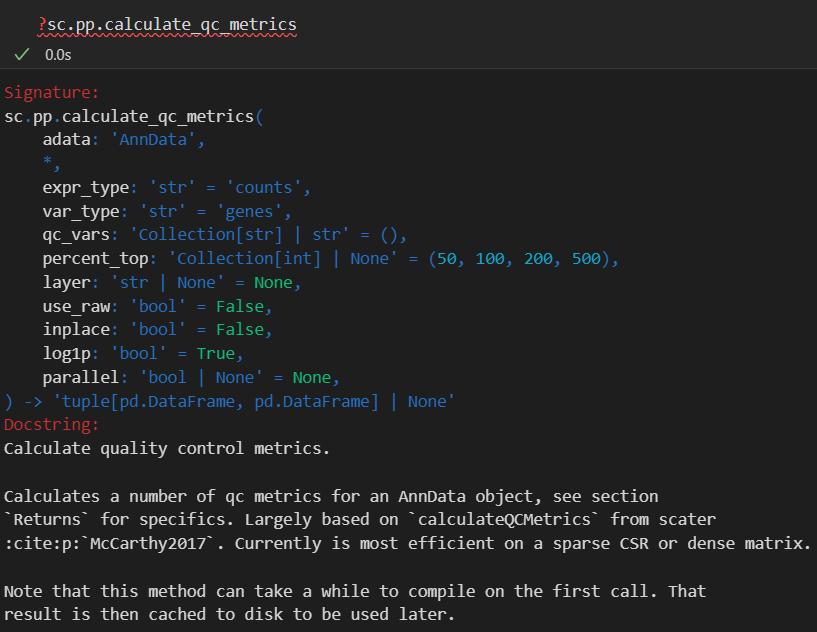
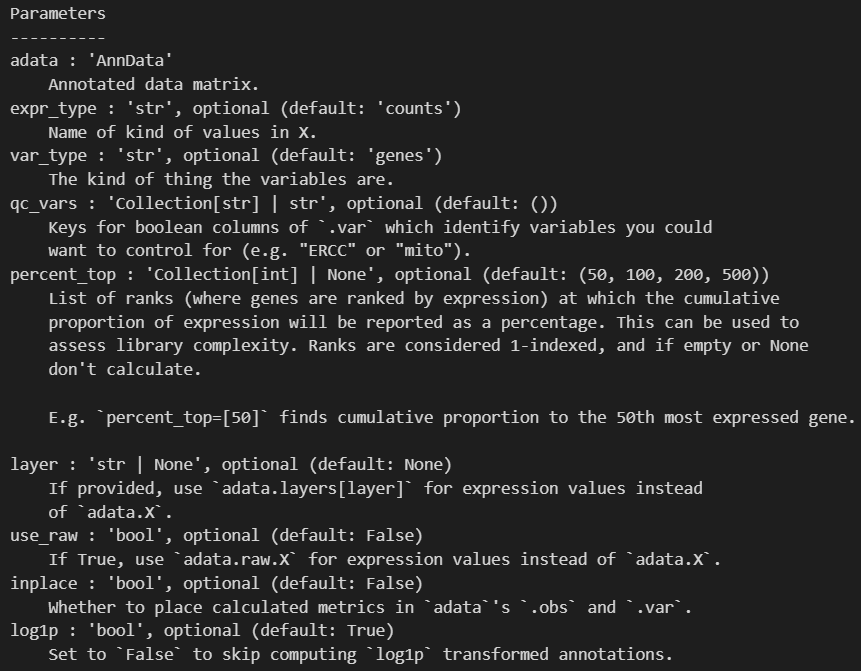
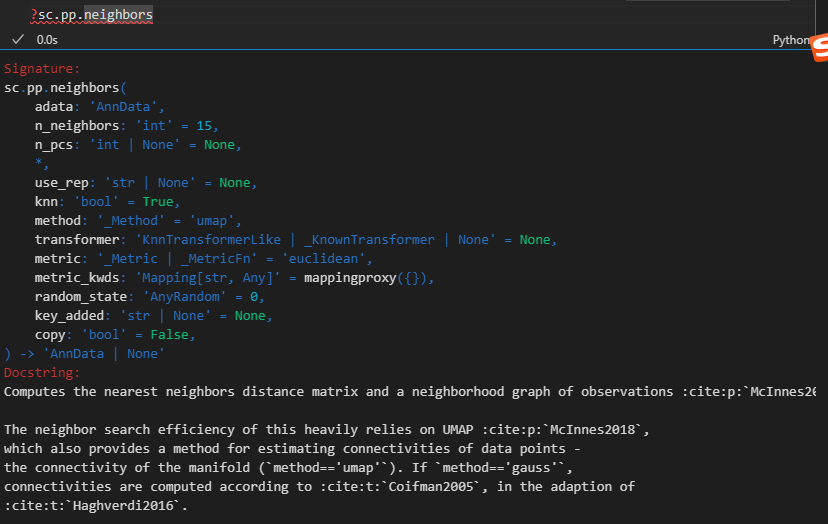
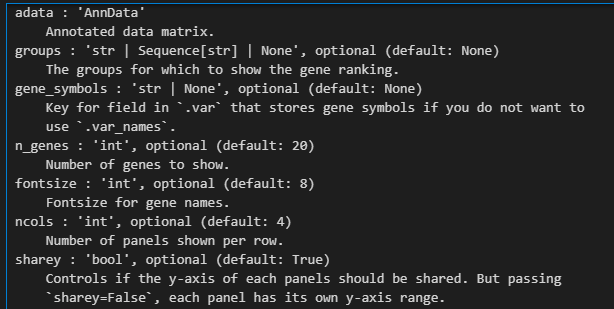
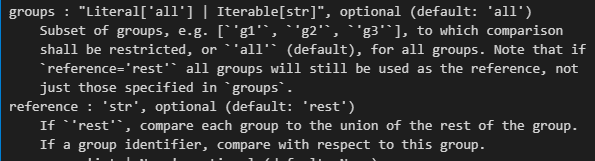
xxxxx上述代码所使用的函数帮助文档如下——,请结合代码详细解释每个参数的意义以及应用
如何理解并解释这个结果,x轴是什么,y轴是什么,比较的是什么指标,图形展示的是是什么意义
参考:
https://mp.weixin.qq.com/s/NjaxcPlivZx7zcweunYPwQ

















 3244
3244

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









