







上次,我们用fastp,Trimmomatic, trim_galore各对测序数据进行了质量控制。
经过质量控制的数据应该要再次进行质量检测,在这里我们不分享这步,有兴趣的朋友可去5.windows Ubuntu 子系统,测序数据质量检测。-CSDN博客。
如果我们得到的测序数据还有宿主序列的话,就需要去除宿主。
我们这里使用bowtie2去宿主。
#bowtie2官网
http://bowtie-bio.sourceforge.net/bowtie2/manual.shtml
# 可以使用conda安装
conda activate dna
conda install -c bioconda bowtie2 -y
conda install -c bioconda samtools -y #sam转bam并排序。
也可以wget下载,再进行环境配置。
bowtie2 -h #出现技术文件说明即是可以使用。
samtools --help #出现技术文件说明即是可以使用。
因为我们下载的数据是从人尿液身上提取的微生物,去宿主之前,首先要下载人基因组。
http://hgdownload.soe.ucsc.edu/goldenPath/hg38/bigZips/ 官网下载hg38和MD5sum.txt
touch md5.txt
vim md5.txt #可以检查HG38的语句。
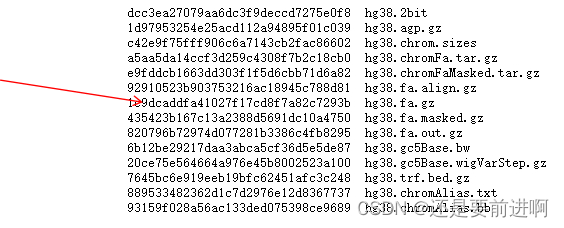
1c9dcaddfa41027f17cd8f7a82c7293b hg38.fa.gz #保存。 md5sum -c md5.txt #ok就代表没问题。 gunzip hg38.fa.gz #解压缩
bowtie2建立索引。
bowtie2-build hg38.fa hg38
最终生成 hg38.1.bt2 , hg38.2.bt2, hg38.3.bt2, hg38.4.bt2 , hg38.fa, hg38.rev.1.bt2, hg38.rev.2.bt2这几个文件。
# bowtie2比对 ,我们进行一下批量比对。
ls *_1_clean.fq.gz > sample.txt
sed -i "s/_1_clean.fq.gz//g" sample.txt
sample.txt中含有的是各个fq.gz文件的样本号。
cat sample.txt | while read id; do
bowtie2 -p 10 -x /mnt/h/db/bowtie2.db/hg38 -1 "${id}_1_clean.fq.gz" -2 "${id}_2_clean.fq.gz" --un-conc-gz "nohuman/${id}.nohuman.fq.gz" | samtools sort -O bam -@ 2 -o "nohuman/${id}.bam"
done
这样就可以进行批量运行了。
1.cat sample.txt: cat 命令用于连接文件并输出到标准输出。在这里,它用于显示 sample.txt 文件的内容。
2.|: 管道符号,用于将一个命令的输出传递给另一个命令的输入。
3.while read id; do ... done: 这是一个循环语句,逐行读取 sample.txt 的内容,并将每行内容赋值给变量 id。循环会执行 do 和 done 之间的命令,直到处理完所有行。bowtie2 -p 10 -x /mnt/h/db/bowtie2.db/hg38 -1 "${id}_1_clean.fq.gz" -2 "${id}_2_clean.fq.gz" --un-conc-gz "nohuman/${id}.nohuman.fq.gz": 这部分是 Bowtie2 的命令,用于进行比对。具体参数含义如下:-p 10: 使用 10 个线程并行处理。-x /mnt/h/db/bowtie2.db/hg38: 指定 Bowtie2 索引文件的路径。-1 "${id}_1_clean.fq.gz" -2 "${id}_2_clean.fq.gz": 指定待比对的 paired-end 测序数据文件。--un-conc-gz "nohuman/${id}.nohuman.fq.gz": 将无法比对到人类基因组的 reads 输出到指定文件。
4.|: 再次使用管道符号,将 Bowtie2 的输出传递给下一个命令。
5.samtools sort -O bam -@ 2 -o "nohuman/${id}.bam": 这部分是 Samtools 的命令,用于对 Bowtie2 的比对结果进行排序和转换为 BAM 格式文件。-O bam: 指定输出文件的格式为 BAM。-@ 2: 使用 2 个线程并行处理排序。-o "nohuman/${id}.bam": 指定排序后的 BAM 文件保存路径和文件名。
bowtie2还有很多参数,需要根据需要调整,建立一个流程并不难,难的是需要,比对结果,根据需要调整参数。















 916
916











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









