






滤食性牡蛎体内巨大的病毒多样性
A remarkably diverse and well-organized virus community in a filter-feeding oyster

Research,2022年1月7日,Microbiome,[IF 16.837]
原文链接:https://doi.org/10.1186/s40168-022-01431-8
第一作者:姜敬哲、方艺菲
通讯作者:姜敬哲、段明、张殿昌
主要单位:中国水产科学研究院南海水产研究所、上海海洋大学、中科院武汉水生所、广东美格基因科技有限公司等
- 摘要 -
背景:病毒在海洋生态系统中发挥着重要作用,许多有关海洋环境病毒的研究已经发表,但栖息在海洋动物中的病毒在很大程度上被忽视了。牡蛎是沿海生态系统中的关键物种,作为滤食性双壳类动物,它们具有群体聚集和固着性的生活方式,目前尚不清楚它们在海洋病毒传播和近岸海洋微生物群落调节中发挥了什么作用。
结果:本文报告了一个牡蛎病毒组数据集(DOV,Dataset of Oyster Virome),包含了728,784个非冗余病毒操作分类单元 (vOTU,≥800 bp)和3,473个高质量病毒基因组,首次全面描述了香港牡蛎体内的DNA和RNA病毒群落。对多维度数据的分析结果显示,牡蛎体内拥有巨大的病毒多样性,并且它们与海洋或其它栖息环境中的病毒非常不同。在牡蛎中还发现了高度多样的圆环病毒(circovirus),表明牡蛎是圆环病毒的潜在热点宿主。值得注意的是,牡蛎中富集的病毒群落并不是随机的,而是组织有序的,病毒群落的组成会对宿主健康状态和外部环境变化做出响应。
结论:绘制的首个牡蛎的病毒组 “图谱”使得牡蛎相关病毒的数量增加了数万倍。研究结果表明,牡蛎提供了一种不同于海水的独特栖息环境,并突出了滤食性双壳类在海洋病毒资源挖掘中的重要性,以及它们在调节海洋生态系统中必不可少但尚不可见的作用。
关键词:香港牡蛎,双壳类,软体动物,宏基因组,圆环病毒,病毒样颗粒富集,多重置换扩增
- 背景 -
作为地球上数量最多、多样性最高的生命体,病毒可感染所有的细胞生物。它们在宿主疾病、代谢、生理和进化中扮演关键角色,影响着海洋生物地球化学循环并塑造地球微生物圈群落。非培养依赖的下一代测序技术最近被用于从多种样本中探索病毒的巨大多样性。在这些发现中,海洋病毒(主要是海洋细菌的噬菌体)的发现进展特别令人印象深刻,包括创建了全球海洋DNA病毒组2.0数据集(GOV 2.0),其中包含从全球收集的145个海水样本中检测到的195,728个病毒种群。
以往许多研究都集中在海水中的游离病毒上,而海洋动物中的病毒在很大程度上被忽视了。海洋动物体内充斥着病毒,它们栖息在宿主的体表、体腔和血液中,与宿主形成了有机整体,参与宿主体内和体外的生命活动。软体动物门的双壳类(如牡蛎、贻贝、扇贝和蛤蜊)是数量庞大的一个动物类群,是海洋生态系统的重要参与者。许多双壳类还是重要的渔业和水产养殖品种,也是气候变化下研究海洋酸化、生物矿化和沿海环境适应的模式生物。一些固着性的双壳类动物,如牡蛎和贻贝,为近岸海洋环境提供了稳定和持久的保护。然而,它们的生活方式,包括高密度的群落聚集及其它海洋生物对牡蛎礁的紧密依存,或许可为海洋病毒传播提供条件。重要的是,作为滤食性动物,双壳类动物每小时可以通过鳃过滤高达5L的水体,从而将悬浮的微生物和颗粒物浓缩到海水中浓度的1000到10万倍。事实上,牡蛎的鳃或肠道组织中富集的人类肠道病毒和mimiviruses的就是这一滤食行为的结果。
双壳类具有半开放的循环系统,其血淋巴被泵入血腔后,使得多种物质得以直接在血淋巴和体细胞之间交换。因此,双壳类动物体内的微生物群落是独特的。以往的研究表明,牡蛎体内的微生物群主要受外界环境扰动影响,但牡蛎体内的微生物群落与水环境中的微生物群落却存在显著差异。这表明牡蛎的内部环境对其宿主微生物群落具有选择性。迄今为止,关于牡蛎中病毒微生物群落的研究很少。双壳类动物究竟为病毒提供了怎样的环境,非常独特还是与海水相似,以及它们是否传播病毒,是否参与调节近岸海洋微生物群落,这些都是有待回答的重要问题。
牡蛎,广泛分布于全球的潮间带,是世界上产量最高的海水养殖品种。我国是最大的牡蛎生产国,占世界总产量的85.3%(FAO,2019年)。本研究中报告了第一个牡蛎病毒组数据集(DOV),包括我国南部沿海养殖最多的牡蛎品种——香港牡蛎(Crassostrea hongkongensis)的不同组织部位、养殖区和采样时间。我们使用病毒样颗粒(VLP)富集和靶向扩增策略,针对性解析了牡蛎RNA和DNA病毒群落的组成、功能及其影响的因素。
- 样本采集及分组 -
本研究的牡蛎样本均为成体香港牡蛎,样本采集时间从2014年6月到2019年7月横跨5年。首先,我们根据采样的时间顺序将样本分为9个时间批次(表S1: Time_Batch_ID, Sampling_Date)。此外,这些样本还有4种分组方法:①扩增分组:根据扩增方法分为全基因组扩增(whole genome amplifcation,WGA)、全转录组扩增(whole transcriptome amplifcation,WTA)、逆转录+WGA(reverse transcription and WGA,RT-WGA),和双链DNA扩增(dsDNA)(表S1:扩增方法)。②组织分组:基于组织来源进行分组,即混合固体组织和流动的血淋巴(表S1: Tissue_Origin)。③站点分组:基于采样的养殖区(BH、HD、LJ、SZ、TS、YJ和ZH)进行分组(图D)(表S1: Sampling_Site)。④健康状况分组是基于牡蛎的健康状态进行分组(即健康或濒死)(表S1: Health_Status)。"健康"指的是在取样之前或之后养殖区的牡蛎没有发生大规模死亡,且样品均为健康个体。“濒死”表明采样时发生了大规模死亡,收集了依然存活但濒死的个体。综上,我们总共用35份动物样本(表S1: Sample_ID)构建了54个病毒组测序文库(表S1: Library_ID)。
- 结果与讨论 -
1. 牡蛎病毒组数据集(DOV)概览
Overview of the Dataset of Oyster Virome (DOV)
采集了35份来自香港牡蛎的混合组织或血淋巴的样本,这些样本来自中国南部沿海7个主要牡蛎养殖区的9个时间点(图1;表S1)。采用三种主要扩增方法(WTA、WGA和RT-WGA)构建了54个牡蛎病毒文库,并进行测序(表S1)。组装后共获得3,347,421个非冗余contigs(≥800 bp)。其中728,784条序列(vOTUs)(21.77%)经BLAST比对被注释为病毒来源(Fig.1A),统称为DOV。它们主要来自于测序深度较高的血淋巴样本(RT-WGA文库)(Fig.1B),而WTA文库中的vOTU数是所有文库中最低的。
值得注意的是,基于的不同参考数据集进行病毒reads的占比分析(mapping rate)(Fig.1E)。当使用从头组装的vOTUs作为参考数据集进行比对时,有平均值高达29.81% reads被鉴定为病毒来源的reads,远高于RefSeq(NCBI)3.50%和RefSeq+GOV+IMG/VR的12.06%的注释比例 (Fig.1E; Table S1)。vOTUs的高注释比例说明我们的VLP富集方案是有效的,且牡蛎中存在着丰富的病毒;而公共参考数据集的低注释比率则意味着在牡蛎中发现的病毒在很大程度上是未知的新病毒。
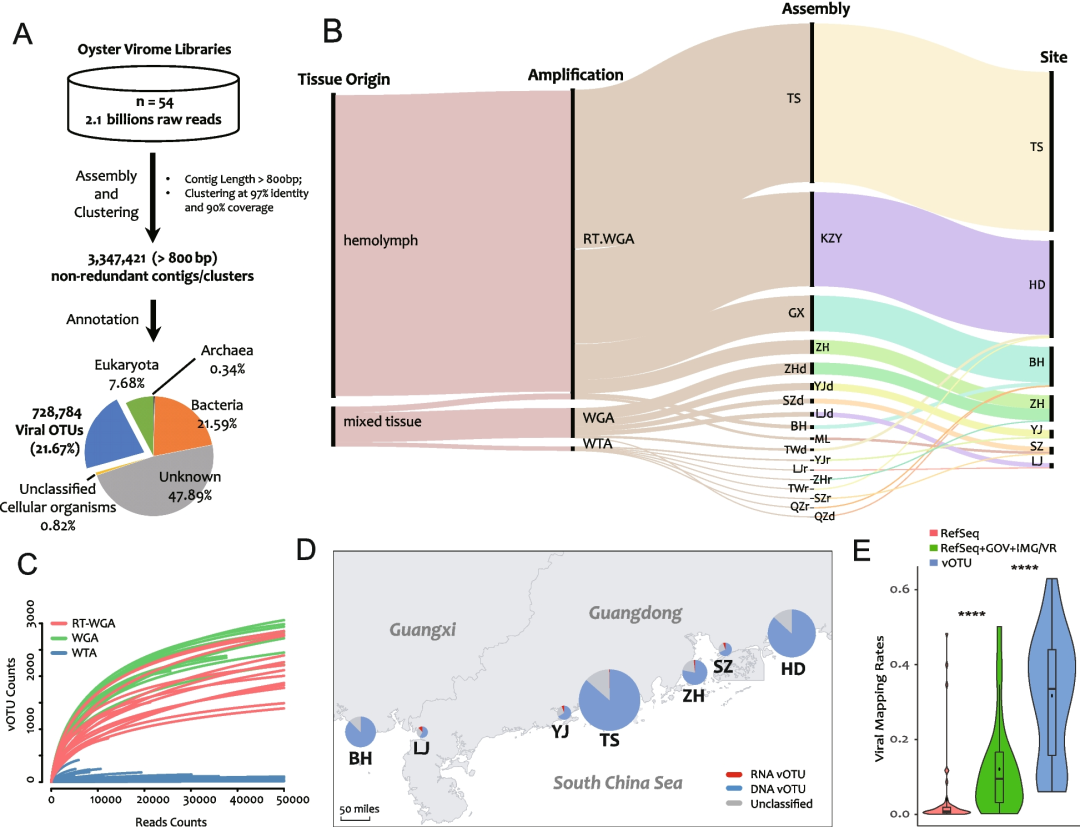
Fig.1 牡蛎病毒组数据集Dataset of Oyster Virome(DOV)概览
A 从头组装和注释分析流程。
B 不同批次和分组之间关系的桑基图。黑色垂直条的高度按比例表示每个分组组装的vOTU的数量。
C 牡蛎病毒文库的稀释度曲线。
D 采样站点分布图和从每个站点检测到的vOTU数量。饼图表示DNA、RNA和未分类的vOTU的数量占比。
E 基于不同参考数据集注释得到的病毒reads在总reads中的占比情况。
2. 牡蛎中的病毒种类
Viruses in oysters
在此之前,牡蛎相关病毒研究主要集中对转录组数据中RNA病毒的挖掘上。Rosani等从C.gigas和C.corteziensis的公开转录组数据中组装出了26个新的几乎完整的RNA病毒基因组。Zhang等报道了4个来自C.gigas的新RNA病毒基因组片段。另有33种新型RNA病毒是从混合双壳类组织样本(其中包括两种牡蛎,C.hongkongensis和C.ariakensis)中鉴定出来。为了充分挖掘牡蛎RNA病毒,本研究构建了33个相关文库(包括19个WTA文库和14个RT-WGA文库)(表S1),但我们只得到了4,958个RNA vOTUs,仅占DOV总数据的0.68%,且全部被归类为未知Riboviria(Fig.S1)。
牡蛎疱疹病毒是研究的最深入的、也是目前唯一已知的双壳类DNA病毒病原。与DOV中多样性较低的RNA病毒相比, DNA病毒在所有DOV文库中占主导地位 (Fig.1)。DOV中丰度排名前三名的病毒科分别是Siphoviridae(28.5~30.61%)、Podoviridae(13.46~42.52%)和Myoviridae(18.36~29.61%)(Fig.2A–C)。尽管MDA方法对环状ssDNA基因组有很强的偏好性,但DOV中Microviridae和Circoviridae的占比仅为2.23%(Fig.S1),这意味着它们的多样性或许低于2.23%,远低于DOV中dsDNA病毒的多样性,说明dsDNA病毒是牡蛎微生物的重要组成。
由于BLAST鉴定病毒的精度有限,大部分(>30%)不能在科水平上进行划分(Fig.S1)。鉴于此,我们使用PhaGCN对8,760条大vOTUs(≥10 kb)进行分析,成功分类了其中的6,362条(Fig.2B),远超了vContact2的214条的分类结果 (Fig.2C),未注释vOTUs的百分比也下降到了11.46%(Fig.2D)。印象深刻的是,在vConTACT2网络图中,DOV节点(vOTUs) 与多数的RefSeq节点均无法聚类,占总节点的74.58%,而RefSeq节点仅占25.42%(Fig.2E),这再次说明了牡蛎体内病毒的独特性。
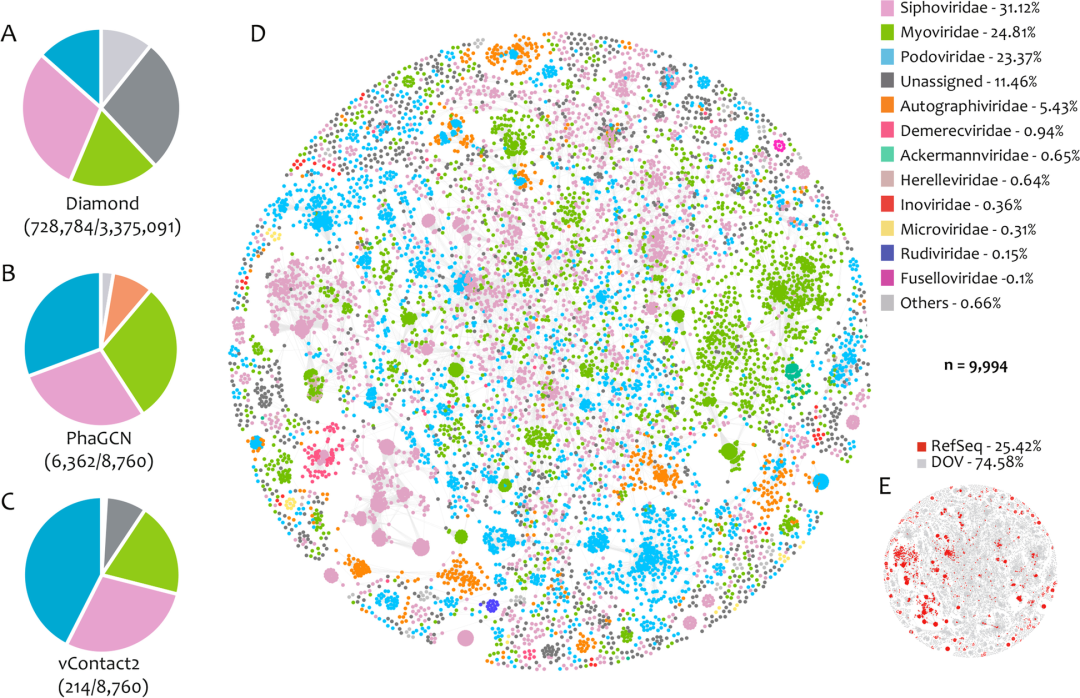
Fig.2 DOV 数据集在科水平上的分类信息
A–C 饼图显示了用不同工具鉴定的各病毒科的占比情况。
D, E 由长度超过10 kb 的vOTUs和NCBI RefSeq病毒组成的vContact2网络图。节点的颜色表示不同的PhaGCN科(D),或者它们的来源(E)。n表示vContact2网络中的节点总数,D和E中各科或来源的百分比列在相应图例后面有标识。
3. 高质量病毒基因组
Near-complete viral genomes
本研究共鉴定出3,473个基因组完整性大于90%的病毒vOTU,其中包括27个RNA病毒基因组(Figs.3和S2; Table S2)。基因组长度为1,206 ~ 60,277 bp,GC含量为24.74~65.70%(Figs.S2)。编码蛋白与已知病毒蛋白的最大一致性为0-93.10%,但主要集中在20-40%范围内(Fig.3; Table S2),这表明绝大多数基因组是新的病毒,在科水平上被判定为未知或未分类 (Fig.3中的灰点) (Table S2)。有科分类信息的病毒至少属于11个DNA病毒科,其中Caudovirales病毒目中包括Podoviridae(173)、Sipoviridae(136)、Myoviridae(66)和Myoviridae(46)(Fig.S2)。但Cirlivirales(圆环病毒为主)和Petitvirales(微小病毒为主)两个目的成员数量最多,分别占可分类基因组的11.27%(396个)和6.98%(240个)(Fig.3; Table S2)。
在科层级上确认的病毒中,“Cruciviridae”、Genomoviridae、Parvoviridae和Circoviridae具有感染动物甚至人类的潜能。红火蚁是目前Cruciviridae唯一已知的宿主;Genomoviridae病毒则可被多种动物携带,包括人类、水豚、乌龟、鸟类和许多其它陆生动物;已知可被Parvoviridae病毒感染的动物则包括有海星、牡蛎、对虾、海豹、人和鸽子;猪和鸟是圆环病毒的易感宿主,此外在几种鱼类、鲸鱼和人类中也报道过圆环病毒。值得注意的是,上述反复出现的潜在鸟类病毒提示我们,鸟类粪便或许是海洋病毒的潜在重要来源,而牡蛎可能是这些病毒的重要储存库和传播热点。
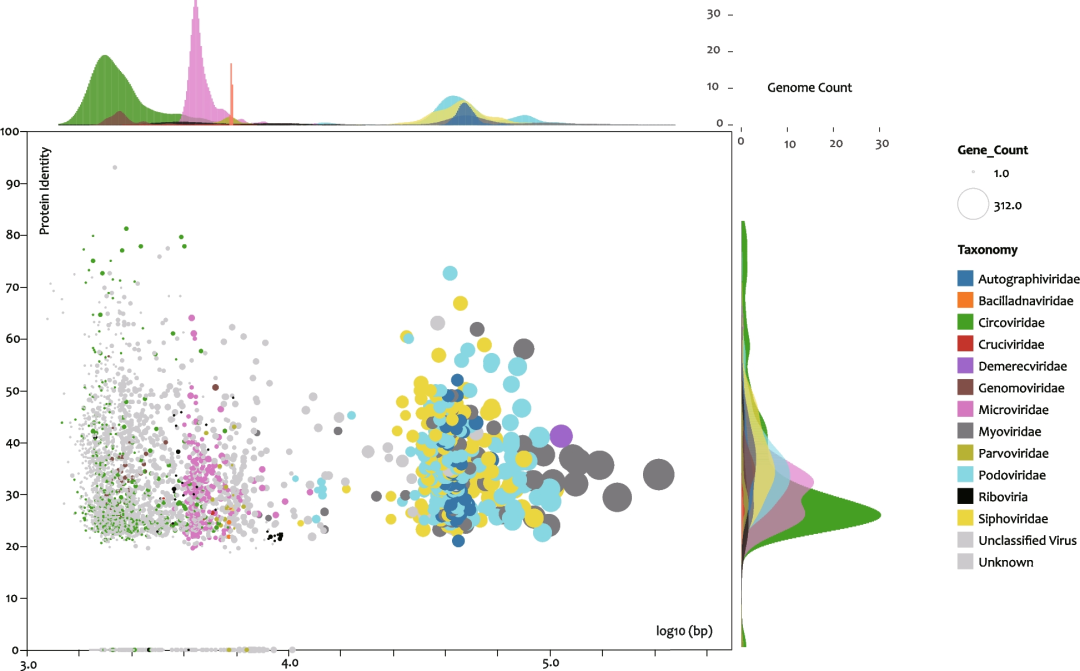
Fig.3 牡蛎病毒组数据集(DOV)中完整和接近完整病毒基因组的散点图
X轴,病毒基因组长度的log10值;Y轴,DOV基因组和已知病毒蛋白序列的一致性。圆圈的直径表示基因组中的ORF数量;颜色表示病毒科水平注释结果。平行于X轴和Y轴的密度直方图显示了相应病毒科的计数分布。
4. 牡蛎相关圆环病毒
Oyster-related circoviruses
在一些病毒组研究中,特别是使用MDA方法的研究中,经常发现Circovirus-like基因组。然而,这些研究多关注的是环境或粪便样本,这意味着很难判定这些Circovirus-like序列的确切宿主。如图所示(Fig.S2),牡蛎相关圆环病毒相关分支广泛分散,并与未注释的分支混合,这意味着许多假定的圆环病毒相关分支尚未被注释。目前已知的所有圆环病毒宿主都属于两侧对称动物(Bilateria),表明DOV中圆环病毒的宿主可能是牡蛎或与牡蛎相关的微型动物。尽管圆环病毒的变异很快,类似于RNA病毒的特性,但在单一一种动物体内中发现如此多的圆环病毒是非常出乎意料的。
圆环病毒是目前已知的最小的动物病毒之一,最早在猪上发现,严重危害畜禽养殖业发展。圆环病毒具有非包膜二十面体结构(直径12-27纳米),其基因组主要编码复制启动蛋白(Rep)和衣壳蛋白两个基因。我们以国际病毒分类委员会(International Committee on Taxonomy of Viruses, ICTV)收录的圆环病毒科成员的Rep序列作为查询序列,通过BLASTP迭代比对,分别从NCBI和DOV中比对出1390条和8763条接近完整的环状病毒相关Rep序列。全部Rep序列的相似性聚类网络图(Fig.S3)表明,环状病毒序列分散在非常多样的聚类群。除了ICTV认定的两个属:Circovirus和Cyclovirus外,其它全部聚类群都属未分类病毒(Fig.S3)。其中来自DOV的序列占比高达86.3%(是NCBI库的6.3倍),广泛存在于所有聚类群中(Fig.S3)。
我们还利用与Circovirus和Cyclovirus聚类在一起的Rep序列(Fig.S3)构建了系统发育树(Fig.4)。结果表明,DOV的大部分Rep序列位于一个独立的分支上,与Circovirus和Cyclovirus分支分离,并且远离污染序列的分支(排除试剂污染的可能性)。我们认为这些来自DOV的Rep序列代表了圆环病毒科下的一个新的牡蛎或双壳类特异属,初步将其命名为Crasscircovirus(Fig.4)。这些发现表明牡蛎(或者双壳类)可能是圆环病毒的热点宿主。这些圆环病毒究竟是牡蛎的病原还是以共生的形式存在于牡蛎宿主体内,是否会像冠状病毒之于蝙蝠一样跳跃到其它海洋或陆生动物,是值得进一步研究的课题。
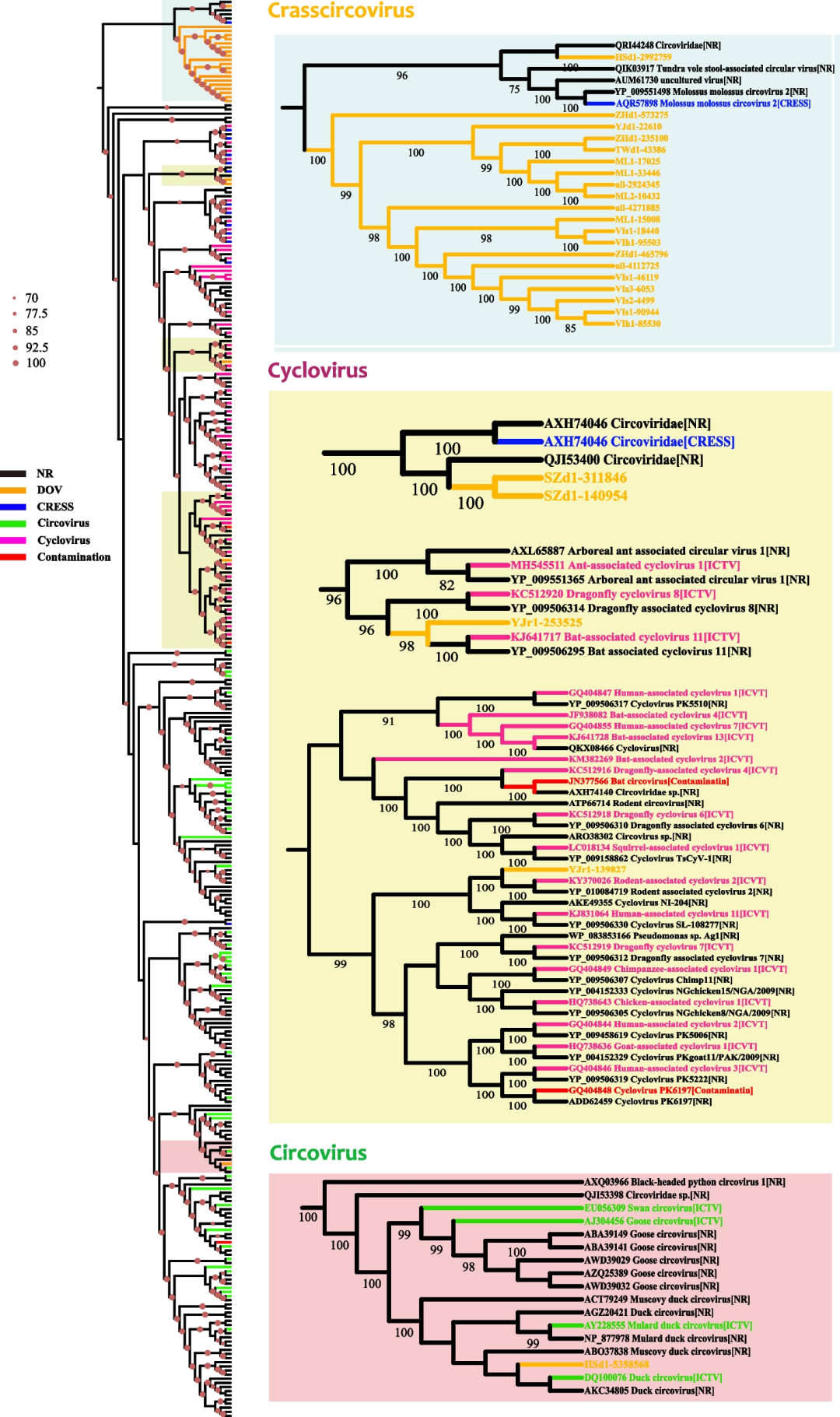
Fig.4牡蛎相关圆环病毒复制启动蛋白 (Rep)的系统发育树
左边的大树显示了国际病毒分类委员会(ICTV)收录的两个标准属以及DOV相关所有Rep的系统发育(Fig.S3)。树枝上红点的直径代表bootstrap值,仅显示超过70的值。右边的小树是左边的树放大的分枝。背景颜色表示不同的病毒属:浅蓝色,Crasscircovirus;黄色, Cyclovirus;粉红,Circovirus。分支的颜色表示数据来源:橙色, DOV;蓝色, Porter等报道的CRESS病毒;绿色, ICTV认定的Circovirus;粉色,ICTV认定的Cyclovirus;红色,Asplund等和Porter等报道的圆环病毒相关污染序列;黑色,其它NCBI序列。
5. RNA病毒与DNA病毒的比较
RNA viruses versus DNA viruses
由于基因组性质不同,以往的病毒组学研究多会将DNA和RNA病毒分开进行定量,比较真实环境中RNA和DNA病毒的多样性及其丰度很有挑战的。本研究使用各种靶向扩增方法(WGA、WTA、RT-WGA),尝试从多个角度比较了同一样本中的DNA和RNA病毒。
首先,研究表明不同的扩增策略可以有效地靶向不同的基因组,因为WTA文库中RNA病毒的vOTUs数量显著高于WGA文库,反之亦然,WGA文库中DNA病毒的vOTUs数量也显著高于WTA文库(Fig.S4A, B)。其次,虽然差异不显著,但WGA文库的α-多样性似乎高于WTA文库(Fig.S4D–F),这与前面的观察结果一致(Figs.1C和S1)。在自然界和公共数据库中,DNA病毒的多样性高于RNA病毒的多样性似乎是普遍现象。然而,DNA病毒多样性比RNA病毒高的结论还需要谨慎求证。因为,RNA基因组极高的突变率会使其序列的鉴定和注释更加困难;此外,RNA基因组的不稳定性和潜在的扩增偏好也将DNA和RNA之间的比较变得更为复杂。
值得注意的是,虽然检测到的RNA病毒的多样性似乎很低,但WTA文库中的总病毒丰度(病毒reads比例)并不输于WGA文库,且明显高于RT-WGA文库中的丰度(Fig.S5A)。值得注意的是,一些WTA文库中RNA病毒的比例高达70%,只检测到很小比例的DNA病毒转录本,展示出与WGA文库中DNA病毒完全不同的丰度比例 (Fig.S5) (Table S1)。因此,这些含有高比例RNA病毒的WTA文库值得进一步分析,以确定里面是哪种RNA病毒在样本中占主导地位,并了解为什么RNA和DNA病毒似乎采用了不同的基因组增殖策略。
6. 病毒群落结构
Viral communities
对病毒群落的研究可以帮助确定牡蛎中富集的病毒是否可以被视为一个有机整体,还是仅仅是一个随机和偶然的组合,以及该群落是否可以对外部影响做出反应。考虑到MDA优先扩增环状ssDNA基因组的偏好,这可能是几个文库中环状ssDNA病毒比例偏高(>80%)的原因(Fig.S5C)。Parras-Moltó等发现基于vOTU的差异排序图显示扩增病毒群落与未扩增病毒群落完美重叠,且与不相关病毒群落分离较强,这表明MDA对病毒群落分析的影响是有限的。
我们首先评估了各种群落参数之间的相关性,包括vOTU数量、病毒reads比例、多样性指数的变化以及测序reads的数量和质量(Fig.S6)。α多样性在三种群落(分别基于RefSeq、vOTU和AMG数据集)之间具有良好的相关性(Fig.S6),这表明几种群落分析的方法得到了互证。其次,正如我们预期的那样,靶向扩增在病毒群落中起决定性的作用(Fig.5A),我们基于参考基因组的群落测定(Fig.5B)进一步证实了这一点。除了扩增方法外,热图(Fig.5C)和F值(Fig.5A)在组织分组之间也表现出显著的差异。即使在半开放的循环系统中,固体混合组织中的病毒群落与液体血淋巴本身的病毒群落也有很大的不同,这说明不同的宿主组织对病毒具有选择性作用。
重要的是,虽然健康状况、采样地点和采样时间对整个群落的影响似乎不如扩增策略和组织来源影响明显(低F值)(Fig.5A),但我们仍然发现所有健康和患病样本之间的αβ多样性都存在显著差异(Fig.S7A, C)。这或许因为疾病引起的免疫力下降导致了宿主体内条件致病菌及其噬菌体的增加。Dupont et al.发现,在疾病爆发期间,OsHV-1 μVar病毒在C.gigas的血淋巴病毒群中占主导地位,会进一步导致病毒多样性低于健康对照组。然而,在本研究的平行组别中,没有检测到死亡组和健康组之间的预期差异(Fig.S9B, C)。
栖息地对宿主微生物群的影响已在许多动物中被报道,环境变化可能是造成差异的原因之一。与自由游动的鱼类不同,牡蛎是固定在礁石上静止不动的,并且每天会过滤大量的海水,对栖息地会有更忠实的响应。本研究确实也发现,地理来源(采样地点)对牡蛎病毒群落有很大影响,同一养殖区来源的样品趋于聚集(Fig.S8)。然而,地理位点的影响似乎弱于采样时间的影响(F值较低)(Fig.5A),这也反映在独特vOTUs的比例上(即仅在一个分组中检测到的vOTUs)(Fig.S9)。时间批次分组中独特vOTU的比例较高,说明病毒群落是随时间动态变化的;而站点之间独特vOTU的比例较低,说明病毒在不同养殖区之间的交换较为活跃。
7. 辅助代谢基因(AMGs)
Auxiliary metabolic genes (AMGs)
在牡蛎体内发现的病毒一来可能是牡蛎的病原,二来可能是牡蛎在滤食过程中随机经过的“路人”,还有一种可能是对牡蛎宿主有互利互惠作用的共生病毒。我们知道,病毒在海洋生态系统的代谢调节中发挥着重要作用。而与海洋病毒类似,我们从DOV中也鉴定出了一定数量的AMGs(9, 091)。他们被注释到12个KEGG category和98个pathway(Table S3)。其中和维生素、氨基酸、能量、碳水化合物代谢相关通路被显著富集(Fig.S10A),这与海水病毒所携带的AMG类型相似。重要的是,AMG群落(Fig.S10B)与vOTU群落(Fig.S10C)具有一致性,丰富度和Shannon指数在两者之间也呈现正相关关系(Figs.S6, S10D, E)。这些结果表明牡蛎的病毒功能与物种群落组成密切相关,虽然很难知道其中的因果关系,但这一结果为我们深入开展海洋动物病毒组功能研究提供了线索。
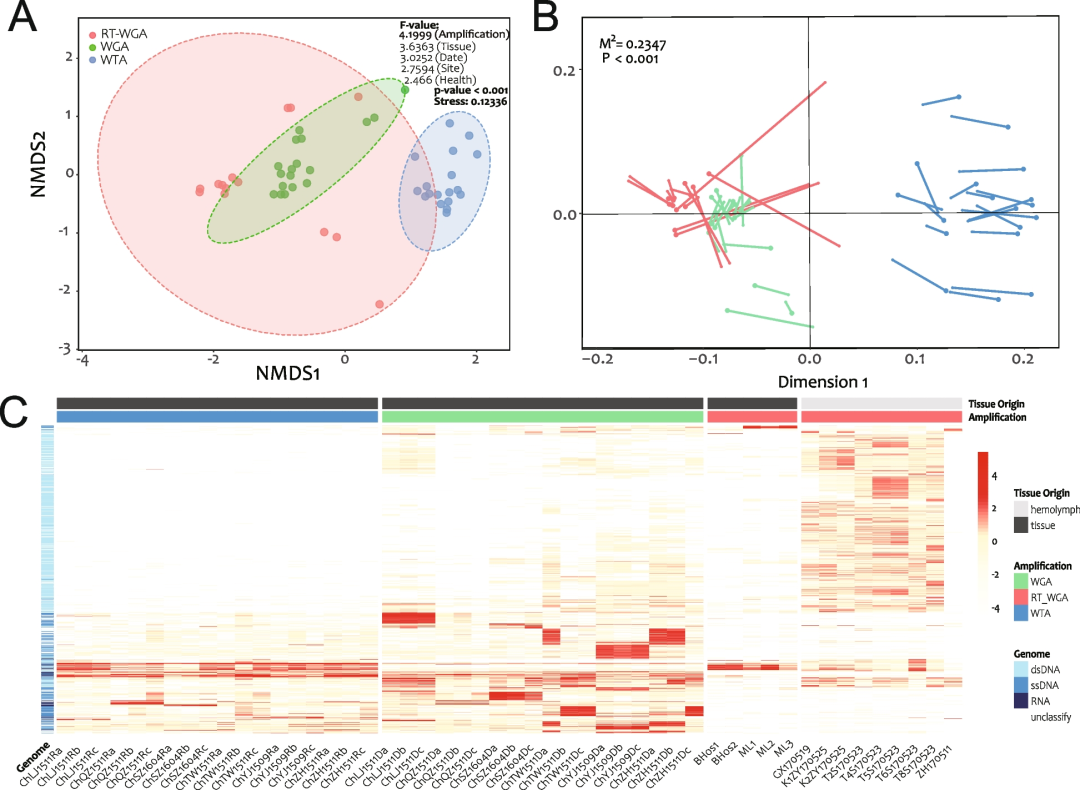
Fig.5 牡蛎病毒数据集(DOV)中的病毒群落
A NMDS分析显示了扩增分组对群落聚类的影响。
B 基于参考基因组(RefSeq, GOV和IMG/VR)和从头组装(vOTUs)比较病毒群落NMDS坐标的Procrustes分析。
C vOTUs热图。vOTUs通过欧几里得方法聚类,并根据病毒基因组类型(dsDNA、ssDNA、RNA和未分类)着色,在Y轴显示;DOV库按扩增策略(WGA、RT_WGA和WTA)和组织来源(血淋巴和混合组织)排序,在X轴显示。
- 总结 -
本研究报道了第一个特定单一海洋动物的病毒组数据集(DOV),为研究和认识海洋中的病毒提供了新的视角。我们从多个方面,包括病毒reads比例、vOTUs、高质量的病毒基因组和圆环病毒相关的Rep蛋白,证明了牡蛎体内蕴含着大量、多样化和独特的病毒类群,牡蛎的滤食性生活方式和高密度栖息环境可能促使其成为海洋病毒的储存库和传播热点。此外,牡蛎体内的病毒群落并不是随机的,而是组织良好的、能够在成分和功能上对宿主组织来源和健康状态以及外部环境的变化做出响应。进一步研究双壳类病毒群落结构和功能,将有助于了解其在近岸海洋微生态调节、疾病传播中的作用,以及挖掘其在沿海生态系统保护和监测方面的潜力。
参考文献
Jiang, JZ., Fang, YF., Wei, HY. et al. A remarkably diverse and well-organized virus community in a filter-feeding oyster. Microbiome 11, 2 (2023). https://doi.org/10.1186/s40168-022-01431-8
- 作者简介 -
第一作者
中国水产科学研究院南海水产研究所
上海海洋大学
方艺菲
2018级联合培养研究生
主要研究方向:贝类病毒的多样性、生态功能与进化
通讯作者

中国水产科学研究院
南海水产研究所
姜敬哲
研究员
渔业生物病害防治研究室副主任,广东省环境功能基因芯片工程技术研究中心主任(兼),中国水产科学研究院“贝类病害与生态防控创新团队”首席专家(PI)。参加工作以来主持包括国家自然科学基金等各类项目总经费超1000万元,以第一或通讯作者身份在Microbiome、Plos Pathogens、Briefs in Bioinformatics、iMeta、Aquaculture等期刊发表学术论文28篇,其中EST高被引1篇,以第一发明人获国家发明专利授权11项,成果转化2项,获得海南省科技进步一等奖1项、广东省科技进步二等奖1项。
猜你喜欢
iMeta简介 高引文章 高颜值绘图imageGP 网络分析iNAP
iMeta网页工具 代谢组MetOrigin 美吉云乳酸化预测DeepKla
iMeta综述 肠菌菌群 植物菌群 口腔菌群 蛋白质结构预测
10000+:菌群分析 宝宝与猫狗 梅毒狂想曲 提DNA发Nature
一文读懂:宏基因组 寄生虫益处 进化树 必备技能:提问 搜索 Endnote
16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流快速解决科研困难,我们建立了“宏基因组”讨论群,己有国内外6000+ 科研人员加入。请添加主编微信meta-genomics带你入群,务必备注“姓名-单位-研究方向-职称/年级”。高级职称请注明身份,另有海内外微生物PI群供大佬合作交流。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍未解决群内讨论,问题不私聊,帮助同行。
点击阅读原文,跳转最新文章目录阅读

















 3355
3355

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









