






自动化和机器学习高通量分离微生物组
High-throughput microbial culturomics
using automation and machine learning

Article,2023-2-20,Nature Biotechnology, [IF 68.16]
DOI:https://doi.org/10.1038/s41587-023-01674-2
通讯作者:Harris H. Wang
- 摘要 -
在微生物组研究领域,细菌纯培养物对于详细的实验和微生物组领域相关机制的研究一直非常重要。传统的从复杂的微生物生态系统中分离单个细菌耗时、费力,且无法将表型和基因型整合起来。在这里,我们描述了开源高通量机器快速分离离株的平台,为满足快速分离的需要。我们发展一种机器学习方法可以将表型和基因联合起来,以便最大化分离微生物多样性并且能够定向分离特定的属。应用该平台,从20个人的粪便样本中产生了个性化的肠道微生物组生物库,包含总计26,997个分离株,占所有丰富类群的80%。对大于 100,000个视觉捕获的菌落的空间分析揭示了共生长模式在瘤胃球菌科(Ruminococcaceae),拟杆菌科(Bacteroidaceae),红椿菌科(Coriobacteriaceae)以及双歧杆菌科(Bifdobacteriaceae),提示了重要的微生物间相互作用。比较分析了生物库中1,197个高质量细菌的基因组,揭示了有趣的个体间及个体内菌株进化、选择和水平基因转移。在许多新兴的微生物组学研究中,更多的努力应被给予应用培养组框架系统的收集和量化分析基于带有高分辨率基因组学信息成像的表型。
- 背景 -
宏基因组学的发展为广泛调查多样的微生物系统的微生物组成提供了有利的工具,包括从土壤微生物到肠道微生物组组成。然而,微生物需要被分离和培养以便探索各种微生物在。依靠“蛮力”随机菌落挑取的传统培养方法是乏味和费力的。提梯度稀释分离微生物的方法使用96孔或384孔板是资源密集型的,这种方法导致重复分离来自同一群体的相同的占优势类群。微流体系统能够在纳升反应器中生长,但克隆分离株很难提取。
考虑到一个典型的微生物组可以包含数百个成千上万的独特物种呈现出长尾丰富分布(也就是说,很少类群是占主导地位,而大多数类群是稀有微生物),通过系统的培养组学进行全面的菌株收集是一个重要而突出的挑战。
微生物可以根据它们不同的表型来区分,如通过它们在特定培养基中的生长能力或是通过代它们产生的谢产物。基于生长的选择可以提高稀有物种的分离率。比如,用含有不同生长素和抗生素的生长培养基来分离。质谱能够被用来鉴定物种,但是这种方法通量低且需要人工处理。随后发展出的成像激活细胞分选技术主要基于多维图像分离真核细胞,但是这种方法需要复杂的仪器而且不能分离细菌。随着近年来人工智能(AI)和深度学习模型训练来辨别多维成像和生物数据的细微特征技术的发展,结合表型和基因组数据的机器学习将改变下一代微生物培养组学。
在这里,我们描述了一个机器学习引导的机器人菌株分离基因分型平台,可以根据需要快速和高通量地生成培养生物库。本系统对比随机选择方法采用智能的,基于成像的算法增加培养组的物种分类多样性。我们演示了本系统的实用性,建立了20名受试者厌氧微生物个性化分离生物库,共得到26,997个分离株包括1,197个微生物高质量基因组草图,跨越394个16S扩增序列变异(asv)。对每个分离的菌株用配对的基因组和形态特征信息,我们训练了一个ML模型,可以仅根据菌落形态预测分类同一性。应用该机器学习(ML)模型可改进靶向分离我们感兴趣的目标微生物。对琼脂平板上的所有菌落进行大规模成像分析,其结果显示了有趣的物种特异性生长模式和物种间的相互作用。全基因组分析个性化的生物库揭示了个体特异性菌株水平的变异和水平基因转移(HGT)的特征在主要的肠道菌群门水平上。
我们进一步发展出了网络开放数据库建设(http://microbial-culturomics.com/)包含由自动培养组学产生的所有分离物的可搜索的基因型,所有分离株的形态和表型数据,作为微生物领域的一个独特的和不断扩大的群体资源。
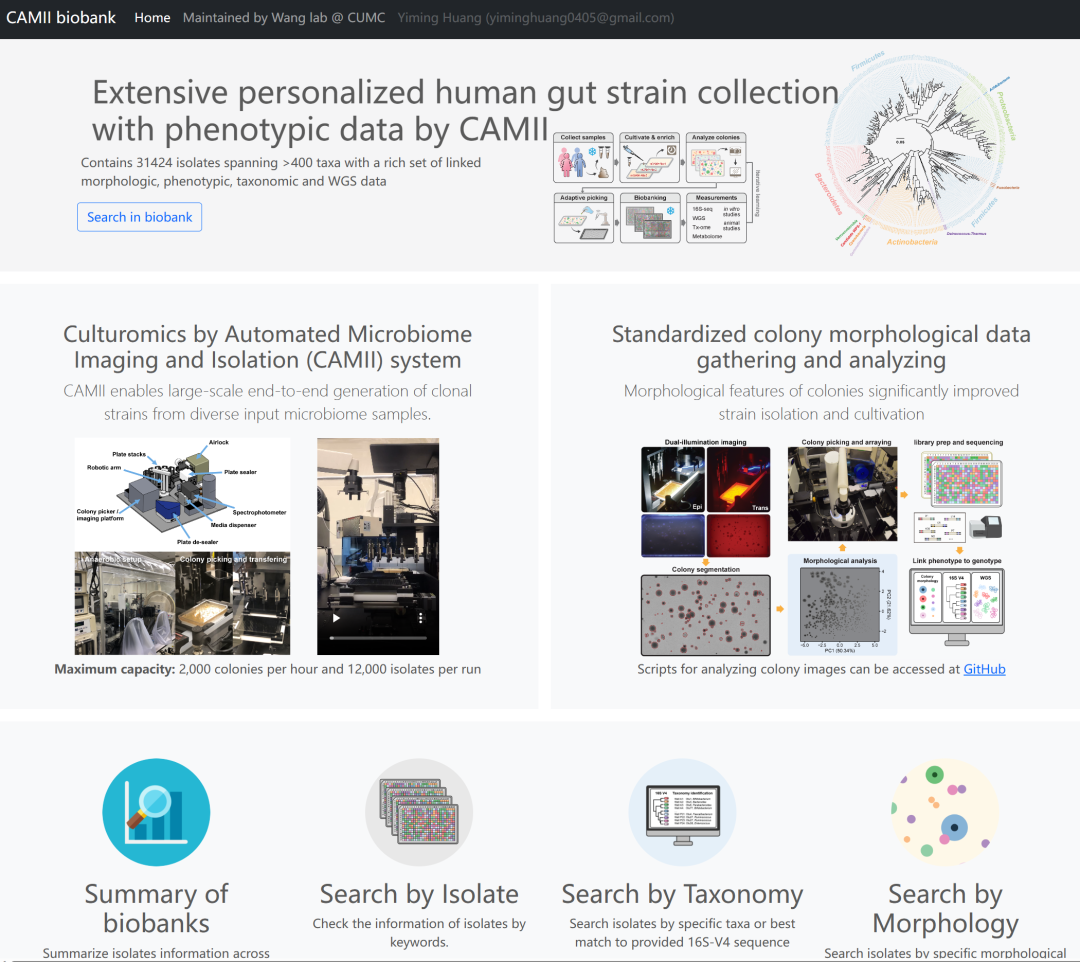
http://microbial-culturomics.com/ 页面截图
- 结果 -
表型数据驱动的培养组和自动化
挑取菌落是一种经典的微生物学克隆分离细菌菌株的方法。菌落在平板上的生长取决于许多因素,包括培养基的成分(例如,可用营养成分)、温度、大气条件(例如,氧合水平)、压力抑制分子(例如抗生素)、pH值、湿度和来自附近菌落的其他可扩散代谢物的影响。根据菌株特异性生理差异,不同的菌株形态可被观察,这些差异往往受细胞形状,硬度,运动和生长动力学,以及色素的产生、细胞外基质和表面活性剂等。虽然这些群体特征很容易量化,但很少有文献记载在在菌株分离期间。因此,利用视觉选择性挑取菌落通常是定性的,而不是标准化的,其结果也会因实验和实验人员不同有很大差异。为了解决这些缺点,我们设计了一个名为微生物组自动成像和分离(CAMII)培养组学平台,用形态学和基因型数据系统化培养组学来分离菌种并进行功能分析。
CAMII平台由四个关键元素组成(图1a),如图所示,讨论如下:(1)采集形态学的成像系统及一个人工智能引导下的菌落选择算法;(2)一个自动的挑取菌落的机器人用于高通量分离及排列菌落;(3)具有成本效益的流水线快速生成挑选的分离物的基因组数据; (4)一个物理分离物生物库和可搜索菌落形态,表型和基因型信息的数字数据库与。因此,这个端到端的培养组学平台能从不同的输入的微生物组中产生分离收集物尽量减少体力劳动。整个成像和隔分离系统使用放置在厌氧室中的现成组件建造,它可以实时控制温度,湿度和氧水平(图1b和补充表1)
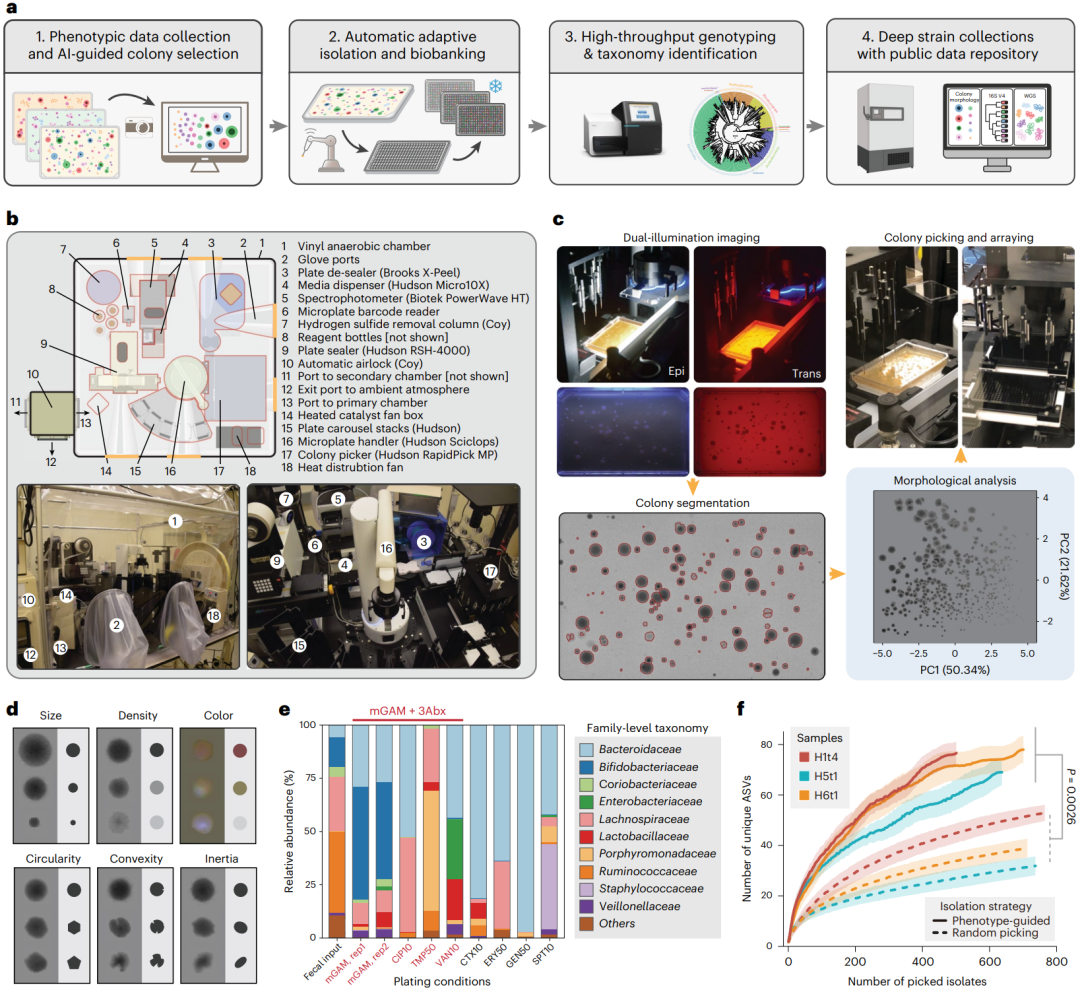
图1. 用表型和形态学数据驱动的微生物分离策略。a、 表型和形态学数据驱动的人类肠道微生物组的分离及数据收集框架。人类粪便样品在不同的抗生素选择下进行铺板和培养,然后对形态多样的菌落进行分离、生物保藏和下游测序分析。b、 自动化厌氧微生物的设置分离和培养系统CAMII。c、 形态引导图解CAMII上的菌落分离。生长在平板上的菌落在透射和落射照明下成像,并进行轮廓分割和形态学特征提取。通过主成分分析对数据进行分析,以确定大多数形态多样的菌落,然后被整合的菌落分离拾取器分离。d、 平板上不同菌落形态的图解。从透射图像中提取菌落大小和形状特征,从落射照明图像中提取菌落颜色特征。e、 粪便样本H1t1与七种不同的抗生素一起培养,以评估它们产生最独特和最多样化的细菌的能力(用16S鉴定到科水平),环丙沙星、甲氧苄啶和万古霉素作为抗生素被选随后的菌落分离。f、 表型引导下获得的独特ASV菌株的分离对比在三种人类粪便样品H1t4、H5t1和H6t1的随机分离。通过CAMII进行菌落分离;进行随机分离在平板上所有检测到的菌落的随机子集上,以表型为导向通过算法对形态学选择的菌落进行分离(补充图1b)。通过对面积进行双侧配对t检验来计算P值曲线下。曲线上的条带表示通过该算法获得的唯一ASV的数量。
CAMII平台具有每小时2,000菌落的分离通量,并且可以处理每次运行12000个菌落,容量是原来的20倍,速度也更快比人工进行菌株分离要好。为确保我们的基因组分析能力匹配机器人分离的通量,我们也开发了一种低成本,高通量测序流程。象杠杆原理,处理液体处理自动化生成16S条形码库rRNA基因测序或全基因组测序(WGS;方法)。在这个流程中,每个分离物的成本是0.45美元,用于菌落分离和基因组DNA (gDNA)制备,16S rRNA测序0.46美元,在Illumina HiSeq平台上,WGS的覆盖范围为> 60×为6.37美元,比商业服务便宜得多(补充表2)。
CAMII平台的一个关键特点是成像系统它收集并学习细菌菌落的形态数据(图1 c)。具体来说,透射光图像,可以显示菌落高度、直径和环状等信息;反射光图片可以显示颜色以及复杂的形态特征比如褶皱,在CAMII数据上捕获的,可以产生多维的和可量化的形态数据集。我们开发了一个定制的菌落分析流程,按照形态特征细分菌落(方法;补充表3和补充图1)。周长和平均半径反映了菌落的大小,而圆度、曲率和惯性反映了菌落的形状。像素强度及其在红、绿、蓝(RGB)通道中的变化突出显示整个菌落的渐变和颜色(图1d)。接下来我们推断形态不同的菌落更有可能是表现出系统发育的多样性,这可以用来提高菌落分离。因此,我们开发了一种成像引导的“智能挑取”策略,基于图像捕捉和选择在空间里最远距离的点代表着最不一样的菌落,通过在多维欧几里得空间中嵌入的菌落来分离更多样化的菌株(补充图1;方法)。为了进一步增加可以培养和检测的细菌的多样性,CAMII也是使用不同的抗生素补充剂,以丰富最独特的和多样化的微生物的不同亚群(补充图2a,b)。例如,在健康人类肠道微生物组样本(H1t1)中,三种抗生素(环丙沙星,Cip;甲氧苄氨嘧啶, Tmp;万古霉素, Van)具有不同的作用机制,引起最明显的富集培养(图1e和补充图2c)。
为了系统地评估成像引导的菌落分离的能力和保真度,我们将CAMII应用于3个人类志愿者肠道微生物组样本(hh14、H5t1和H6t1) (补充表4)。对平板培养的菌落形态学数据进行主成分分析(PCA),评价信息量最大视觉特征(图1c和补充图1c;方法)。有趣的是,菌落密度和大小是最主要的特征(分别是主成分1和主成分2),可以解释72.0%的形态变异(补充图3)。然后使用CAMII机器人分离出6144个菌落,大约有一半的菌落从mGAM平板中随机挑选,另一半则使用我们的成像引导“智能挑取”策略和抗生素选择。分离的菌株放在384孔板中进行培养,并进行16S测序用于分类鉴定。独特的16S的V4区序列聚类分析基本鉴定到种水平。值得注意的是,根据表型数据的菌落分离,要比从三个微生物组随机分离的更加多样化(图1f)。例如,要获得30个唯一的扩增序列变异asv,使用我们的成像选择策略,仅需要分离85±11个菌落即可,而随机选择所需菌落为410±218个。值得注意的是,这种提高了的分离效率在整个挑取过程中保持不变,这意味着在所需的分离深度范围内,使用我们的策略具有持续的优势(补充图4a),并且生成的分离集合更好地代表着潜在输入微生物多样性,并且随后的香农多样性指数也是高的(补充图4b)。
菌株的系统发育分析显示CAMII优化的菌落挑取大大提高了获得的微生物的多样性(补充图5)。鉴于在日益增加的微生物菌株中发现独特的扩增序列变异(ASV)变得越来越困难,这种优势尤为明显。总之,这些实验结果例证了CAMII平台中人工智能引导的数据驱动分离框架能够大大增加培养组的效率,节约劳动力,尤其是对分离稀有的微生物物种。
快速生成个性化肠道微生物分离生物库
虽然来自不同人的微生物组可能共享相似的细菌物种,但属于这些物种的菌株对于个体来讲是高度独特,它们可以在同一个寄主上共存多年。我们试图展示CAMII生成的20个健康人体的个性化的肠道菌群(补充表4和补充图6a、b)。总共对102,071个菌落进行图像分析,并通过16S rRNA基因测序对26997个菌落进行挑取和分类鉴定(图2a),得到394个独特的asv,覆盖了广泛多样的健康共生肠道微生物群(图2b,c和补充表5)。
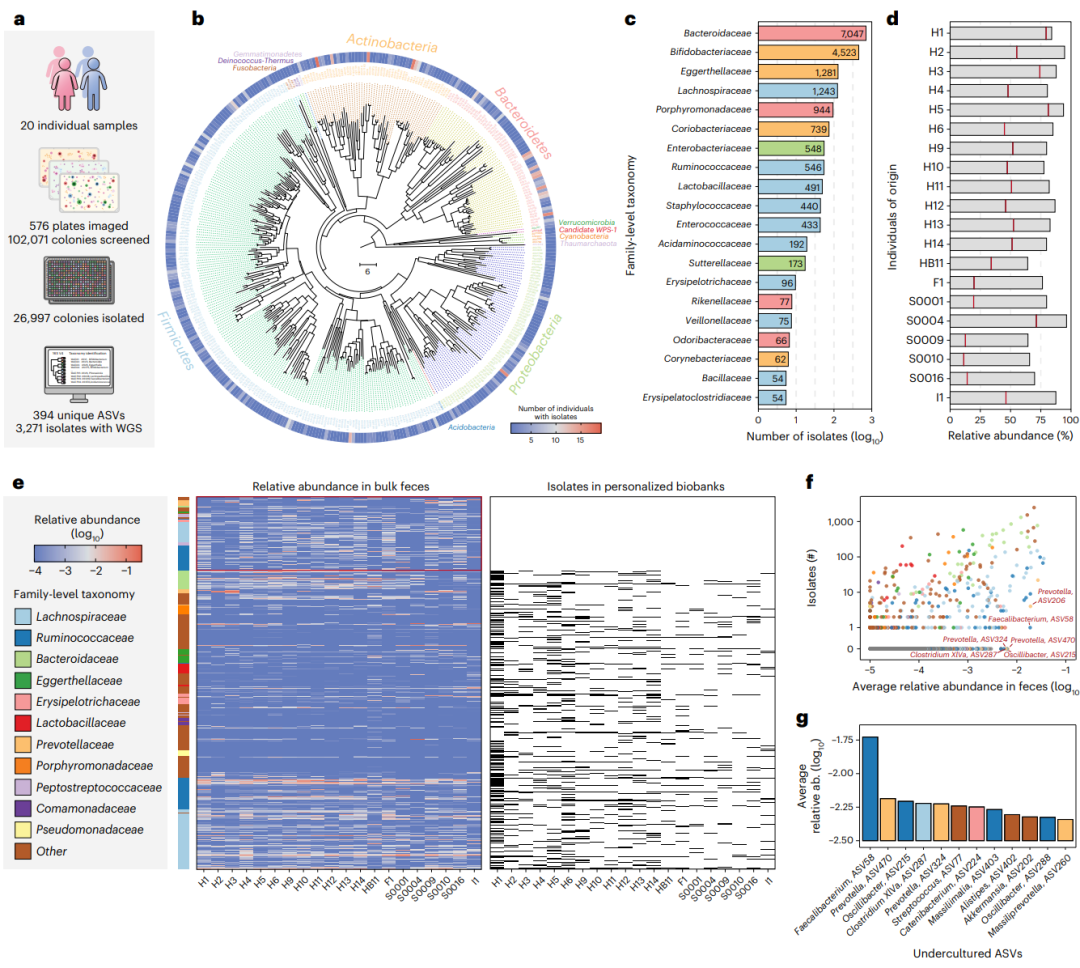 图2. 20个个体生成个性化的肠道分离物生物库。a、 20个个性化肠道分离生物库的统计数据。b、 394个扩增子序列变异( ASV)的系统发育树,覆盖26997个肠道微生物组分离株。以16S rRNA的V4区序列为基础采用邻接法构建了系统发育树。分支颜色区分细菌门,外圈显示在20个生物库中主要分离的ASV。c、 前20个科水平的分离菌株的数量。d、 ASV的累积相对丰度表示为来自原始粪便样本中个性化生物库的分离物。标尺显示从整个集合中的任何个体中分离出来,红线显示分离菌株源自同一个人。e、 丰度相对丰度的热图原始粪便样本中的丰富的ASV的丰度热图以及在生物库中是否存在。平均相对丰度 >0.1%的ASV,左侧栏表示它们的科水平的分类。在个性化生物库中发现的ASV在右侧热图中显示为黑条,在任何生物库中未找到未培养的ASV都会突出显示。f、 原始粪便样本中的ASV平均相对丰和整个收集的ASV分离物的数量相关性。难培养的高度丰富的ASV,也就是说,分离株较少被突出显示。g、 丰度最高但整个集合中有不超过2个以上的分离物的高丰度的ASV的相对丰度。条形图的颜色表示科级分类水平的分类单元。
图2. 20个个体生成个性化的肠道分离物生物库。a、 20个个性化肠道分离生物库的统计数据。b、 394个扩增子序列变异( ASV)的系统发育树,覆盖26997个肠道微生物组分离株。以16S rRNA的V4区序列为基础采用邻接法构建了系统发育树。分支颜色区分细菌门,外圈显示在20个生物库中主要分离的ASV。c、 前20个科水平的分离菌株的数量。d、 ASV的累积相对丰度表示为来自原始粪便样本中个性化生物库的分离物。标尺显示从整个集合中的任何个体中分离出来,红线显示分离菌株源自同一个人。e、 丰度相对丰度的热图原始粪便样本中的丰富的ASV的丰度热图以及在生物库中是否存在。平均相对丰度 >0.1%的ASV,左侧栏表示它们的科水平的分类。在个性化生物库中发现的ASV在右侧热图中显示为黑条,在任何生物库中未找到未培养的ASV都会突出显示。f、 原始粪便样本中的ASV平均相对丰和整个收集的ASV分离物的数量相关性。难培养的高度丰富的ASV,也就是说,分离株较少被突出显示。g、 丰度最高但整个集合中有不超过2个以上的分离物的高丰度的ASV的相对丰度。条形图的颜色表示科级分类水平的分类单元。
为了评估这个分离菌群的全面性,我们通过16S rRNA基因批量测序计算了相应粪便样本中分离asv的丰度(图2d)。值得注意的是,对每个个体而言,80.9±9.4%的asv丰度代表在整个分离的菌株中至少出现一次。来源于每个人的分离菌株占细菌ASV总数的45.6%±21.6%(图2d)。此外,比较分离菌株和粪便标本的批量测序结果,大部分高分度和占优势的asv在收集的菌群中至少被分离一次(补充图6c-e)。此外,每个人的分离收集菌株与粪便样本的微生物组有相似的Shannon’s多样性指数(补充图6f、g)。
总之,我们展示了利用CAMII构建的深度人类肠道菌群分离物集合包含26997个菌株,包括394个ASV,集合了形态学、表型、分类学和基因组(WGS)信息与一体。为了增加其对研究群体的效用,我们进一步开发了一个可搜索的在线资源(http://microbial-culturomics.com)来容纳所有启用CAMII的生物库数据,包括基因组,表型和图像数据。我们预计该门户网站将进一步促进基因型到表型分析,并实现更多来自其他环境的共享菌株的收集。
识别肠道微生物组中未培养的“暗物质”
以前的研究表明,来自不同地区的许多环境微生物环境很难在实验室中培养。因此,我们利用系统生成的分离生物库进行评估人类肠道微生物组的可培养性,并在我们的实验中鉴定仍然难以分离的细菌ASV。在所有20个个性化分离收集群,我们确定大量粪便中是否有丰富的ASV(平均相对丰度 > 0.1%)存在于生物库中。值得注意的是未培养的肠道细菌属于Ruminococcaceae and Lachnospiraceae(图2e和补充表6),以前也被记录为“不可培养”。对于每个ASV,我们比较了在我们总分离收集中产生的分离物数量与它们在粪便中的平均丰度(图2f),结果显示二者之间存在正相关。此外,我们还鉴定了一组高丰度但难以培养的细菌,包括Faecalibacterium ASV-58, Prevatella ASV-470 和 ASV-324, Oscillibacter ASV-215 和Clostridium XlVa ASV-287(图2g)。有趣的是,我们对获得的一个分离物Faecalibacterium ASV-58进行了WGS,与Cibiobacter qucibialis的宏基因组组装基因组(MAG)有超过>98%的相似性。据报道,我们收集的这种菌株,它是人类基因组中最丰富的未培养物种并且在肠炎(IBD)患者中高度耗竭,其他Faecalibacterium菌株一样。
我们进一步通过WGS将我们生物库中的分离株与现有数据库进行了比较,并确定了11个未被在任何参考集合(BIO-ML、CGR和HMP)中培养,但是仅与SGB集合中的MAG关联(补充图7和补充表7)。例如,除了 Faecalibacterium ASV-58, 我们还分离了另一种丰富度很高的菌株Faecaliberaterium 在粪便中,这进一步扩大了可培养的肠道微生物群。总之,这些结果强调了可培养的菌株及基于我们现在的培养基和培养条件还保留了多样的微生物未被培养,这为未来受关注的肠道微生物组中”暗物质“培养组提供指导(补充表6)
目标菌株形态学的分类预测
从微生物组样本中重点培养感兴趣的细菌可能对机制研究至关重要。不幸的是,我们缺乏以特定方式选择性培养大多数细菌物种的能力。因此,挑选大量菌落并依靠统计概率是获得感兴趣菌株的是唯一实践方法。然而,这种策略往往过于消耗资源,可能需要手动挑取数千个菌落。CAMII基于菌落形态的分类鉴定,提供了ML指导的自动化的菌落选择方法,因此在理论上可以加强有目标菌株的分离。为了测试这一点,我们系统地探索了收集深层肠道分离物,分析形态学和基因型数据之间的关系。有趣的是,不同属的群落表现出不同的形态模式(图第3a、b段)。例如,Dorea、拟杆菌和Collinsella的菌落通常较大且菌丝密集,但是表现出不同的圆环(科林氏菌 > 拟杆菌类 > Dorea),反映了它们生长特征的差异。另一方面Faecalibacterium菌落更小、更暗,我们早期的结果也显示它们的可培养性差。此外,菌株的系统发育关系与菌落形态是显著富集的是显著聚集的 = 图3c)PERMANOVA检验结果为0.008。例如,梭状芽孢杆菌的大多数属(Clostridia) 形态学的排序,梭状芽孢杆菌彼此更接近(图3c)。因此,菌落形态可能嵌入大量可能与分类身份相关的信息量。
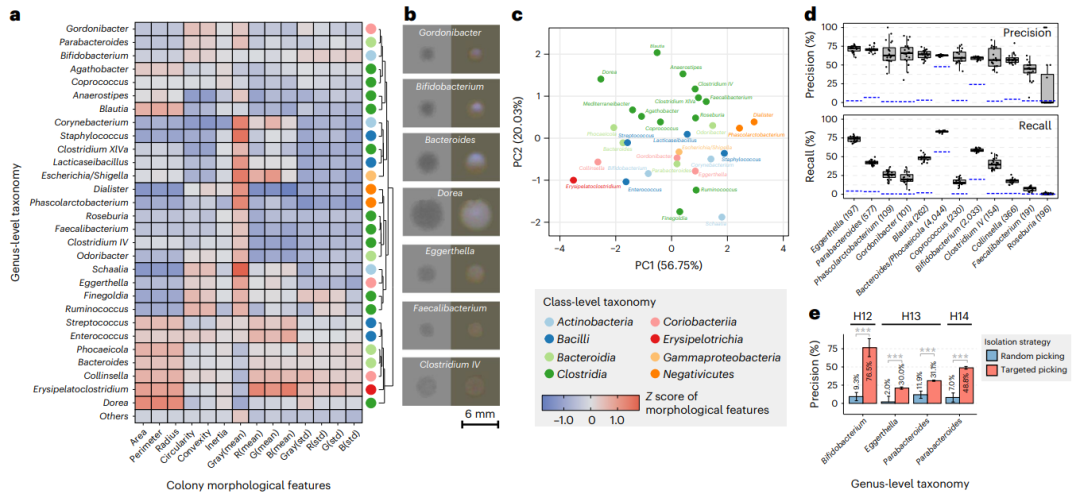 图3. 用菌落形态学预测分类鉴定增强目标菌株的分离。a、 不同细菌属特征形态特征z得分热图。不同的属表现出多样性形态学模式,层次聚类分析划分为不同的组,右边的彩色点表示它们的分类水平。b、 菌落图像示例。透射图像位于左侧;落射照明的图像在右侧。c、 基于其菌落形态特征的属水平的主成分分析排序,颜色表示纲-水平分类。d、 通过随机森林分类基于形态学特征的对细菌属预测。括号中的数字表示每个属的分离株数。模型训练和评估是自举的20次,方框图显示了性能的方差(n = 20). 蓝色线条表示空模型的性能。方框图元素的定义-中线,中线;方框极限,第25个四分位数上下;须状物,1.5倍四分位间距。e、 基于模型的目标隔离的性能。标尺表示单个特定模型的预测精度平均值被引导20次,误差条表示标准偏差。P值通过双侧Student t检验对n的精度进行计算 = 20随机初始化的模型自举。
图3. 用菌落形态学预测分类鉴定增强目标菌株的分离。a、 不同细菌属特征形态特征z得分热图。不同的属表现出多样性形态学模式,层次聚类分析划分为不同的组,右边的彩色点表示它们的分类水平。b、 菌落图像示例。透射图像位于左侧;落射照明的图像在右侧。c、 基于其菌落形态特征的属水平的主成分分析排序,颜色表示纲-水平分类。d、 通过随机森林分类基于形态学特征的对细菌属预测。括号中的数字表示每个属的分离株数。模型训练和评估是自举的20次,方框图显示了性能的方差(n = 20). 蓝色线条表示空模型的性能。方框图元素的定义-中线,中线;方框极限,第25个四分位数上下;须状物,1.5倍四分位间距。e、 基于模型的目标隔离的性能。标尺表示单个特定模型的预测精度平均值被引导20次,误差条表示标准偏差。P值通过双侧Student t检验对n的精度进行计算 = 20随机初始化的模型自举。
我们评估了菌落的分类身份是否可以仅通过将它们的形态学信息结合到平板上来唯一预测。我们从随机选取的一个小的分离物库(占总数的70%;方法,用形态和分类数据,训练了一个随机的森林分类模式,然后对其余30%的分离株进行了模型性能评估。值得注意的是,我们的模型实现了训练数据集大多数属中有100多个分离株的准确度约为70%(图3d)。属水平的召回率差异更大,强调使用更复杂的开放机会模型来学习其他独特的菌落特征。一些属例如Eggerthella具有较高的精确度和召回率,这表明保守和独特的菌落形态可以是特异性的用于分类预测。当分析来自同样的ASV,我们发现菌落形态是高度保守的在来源于同一个人体内分离物,但在相当多的变异发生在不同人群的分离物(补充图8)。考虑到不同的人通常携带相同物种的不同菌株,我们的结果表明菌落形态存在高度的菌株水平变异。
为了评估人工智能告知的菌落特征是否可以提高微生物的分离,我们接下来将随机森林模型应用于我们生物库中的分离数据,是来源于三个不同的人(H12,H13和H14)。该模型用于预测Bifidobacterium, Parabacteroides and Eggerthella菌落,来自同一来源粪便的新的培养平板。然后这些菌落随后通过CAMII和16SrRNA测序以确认分类身份(方法)。值得一提的是,形态学引导下的菌落挑取法显著提高了分离这些目标属的效率,平均高达八倍(图3e),大大提高了挑选菌落的精度,减少了以往的需要筛选许多菌落才能找到所需微生物的需要。这些结果强调了我们的将表型到基因型相结合生物库数据集的价值,并证明了单独从视觉菌落特征来做分类预测,这可以大大增强目标微生物分离。
肠道微生物中的细菌间生长关联
细菌菌落可以通过物种间的相互作用,如争夺营养或交叉喂养必需代谢产物从而影响邻近的其它微生物的生长。先前的研究表明,邻近的细胞可以以可预测的方式严重影响菌落的大小。由于CAMII可以连续跟踪菌落的动态生长,我们系统地探究了肠道微生物菌株在琼脂平板上的的共生长模式。粪便样本(H1t5;补充表4)铺板并进行每日拍照,随后对所有菌落在培养6天时进行菌株分离并用16SrRNA测序鉴定(图4a)对于每种ASV,菌落的累积面积在琼脂平板上与它们在原始粪便中的丰度相关样品(补充图9),表明我们的体外条件通常促进生长到与肠道中相同的程度。令人感兴趣的是,属于粪杆菌属Faecalibacterium的菌落最初生长较慢,只有在其他物种存在的情况下才开始出现附近生长的菌落(图4b;方法)。这一观察结果表明共生或互惠的相互作用可能在存在于粪杆菌属Faecalibacterium和其他物种。
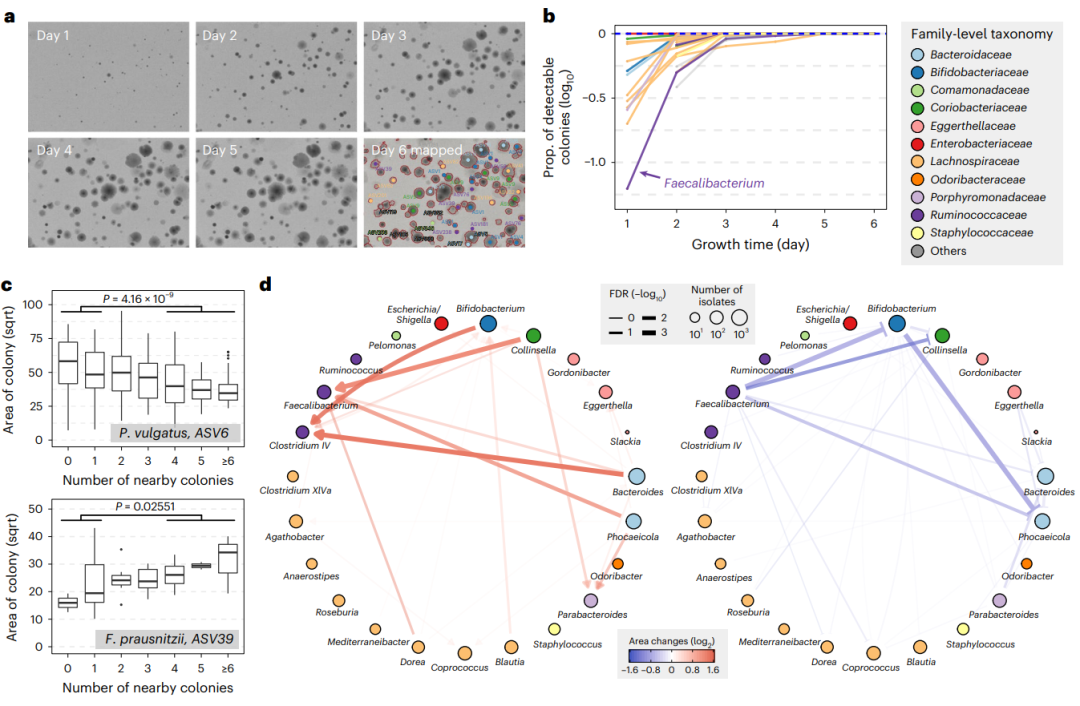 图4. 通过菌落形态绘制肠道微生物群之间的相互作用分析。a、 示例板的图像显示培养6天的菌落特征及 平板上16SrRNA测序结果。b、 不同时间点下的可检测菌落的比例,与每个属的第6天相比。颜色表示科水平的分类。c、 两个代表性的ASV的两个菌落大小和附近菌落数量的相关性,完整的相关性列表见附表8。单侧Mann–Whitney U检验计算P值。附近有不超过一个或不少于四个菌落的菌落(n = ASV-6为101对82,ASV-39为17对9)。箱线图:中心线:中间线;箱子限位:上限和下限四分位数;箱须:1.5×四分位间距。d、 属间配对生长促进和抑制网络。定向生长促进作用如红色尖锐箭头所示;定向生长抑制效果蓝色钝箭头表示。节点代表细菌属,并按科着色。节点大小与本分析中使用的分离株数量成比例,边缘宽度与相互作用的重要性成比例。
图4. 通过菌落形态绘制肠道微生物群之间的相互作用分析。a、 示例板的图像显示培养6天的菌落特征及 平板上16SrRNA测序结果。b、 不同时间点下的可检测菌落的比例,与每个属的第6天相比。颜色表示科水平的分类。c、 两个代表性的ASV的两个菌落大小和附近菌落数量的相关性,完整的相关性列表见附表8。单侧Mann–Whitney U检验计算P值。附近有不超过一个或不少于四个菌落的菌落(n = ASV-6为101对82,ASV-39为17对9)。箱线图:中心线:中间线;箱子限位:上限和下限四分位数;箱须:1.5×四分位间距。d、 属间配对生长促进和抑制网络。定向生长促进作用如红色尖锐箭头所示;定向生长抑制效果蓝色钝箭头表示。节点代表细菌属,并按科着色。节点大小与本分析中使用的分离株数量成比例,边缘宽度与相互作用的重要性成比例。
为了用CAMII更系统地研究物种间的相互作用,我们一起分析了菌落形态、分类地位和菌落邻域数据。我们聚集了102071个可视化捕获菌落的形态数据和物理坐标(26997个分离的),并评估了菌落的生长是否受到相邻区域的影响。令人惊讶的是,我们观察到了一些有趣的可能反映种间相互作用的共生长模式(Supple附录表8)。例如,硫化乳杆菌Phocaeicola vulgatus ASV-6的菌落大小与相邻细胞的数量呈负相关,这与在肠道中P. vulgatus 和其他乳杆菌之间普遍存在的由竞争或拮抗介导的负面的相互作用的情景一致(图4c)。另一方面,Faecalibacterium prausnitzii ASV-39,初始阶段生长较慢,但是由其他菌落存在下,菌落生长会变大,反应了积极的物种相互作用(图4c)。
接下来我们引入了邻近菌落的分类信息来观察一个特定的属的菌落大小如何受到其他属的影响。简单地说,对于每一对属,我们比较了一个属在有另一个属的菌落存在和不存在的情况下对其菌落大小的影响(方法)。值得注意的是,我们从两个属Faecalibacterium 和Clostridium IV中鉴定的分离株,当与Bifidobacterium, Phocaeicola 和 Bacteroide分离物靠近时,菌落变大,生长快(图4d)。据报道,Faecalibacterium 和 Clostridium IV 是肠道中主要产生丁酸的细菌,共培养生长可受益于Bifidobacterium和Bacteroides,这与我们的观察是一致的。另一方面,我们观察到Phocaeicola分离株与Faecalibacterium分离株作为邻居共培养,长势慢,菌落小(图4d),表明共生相互作用可能只对一方有利。此外,与我们先前相关性分析结果一致(检查相邻分离物数量不考虑相邻菌株的身份),Phocaeicola and Bacteroides的生长可以被多个其他属抑制,表明进一步调查以便更好的了解肠道微生物群之间这些积极和消极相互作用的潜在机制是非常必要的。总之,我们的研究结果表明,CAMII可以揭示由种间相互作用指导的菌落共生长模式,这可能有助于鉴定生长促进微生物及它们的能够刺激挑剔的物种生长的可扩散代谢产物。
人群个体内及个体间肠道菌株的基因组多样性
绘制一个人的肠道细菌的菌株水平全基因组多样性对于理解肠道微生物定殖的动力学以及细菌对每个人体宿主的选择和适应的驱动因素非常重要。CAMII系统的一个关键优势是能够对大量分离物进行分离和全基因组测序(WGS),以提供帮助研究个体间和个体内的菌群基因组变异。因此,我们从20个人微生物组生物库中选取了最主要最独特的ASVs,并进行WGS,产生1197个高质量基因组草图(补充图10和补充图表9)。进一步分析基因组组装以确定分离株的准确种级分类(方法)。
我们首先在我们的分离物收集中探索了人际菌株水平的基因组变异(方法)。与先前的报道一致,同一个体内的大多数分离株有很少的基因组变异(即少于102个SNPs),而个体间在全基因组水平上有103-105个SNPs存在差异(图第5a段)。有趣的是,同一物种的一些系统发育显著不同的菌株(即超过104SNP),在同一个人体内共存(图5a)。例如,分离出了两种不同的P.vulgatus菌株来自H4个体和两种不同的B. uniformis 菌株在H2个体中发现(补充图11)。
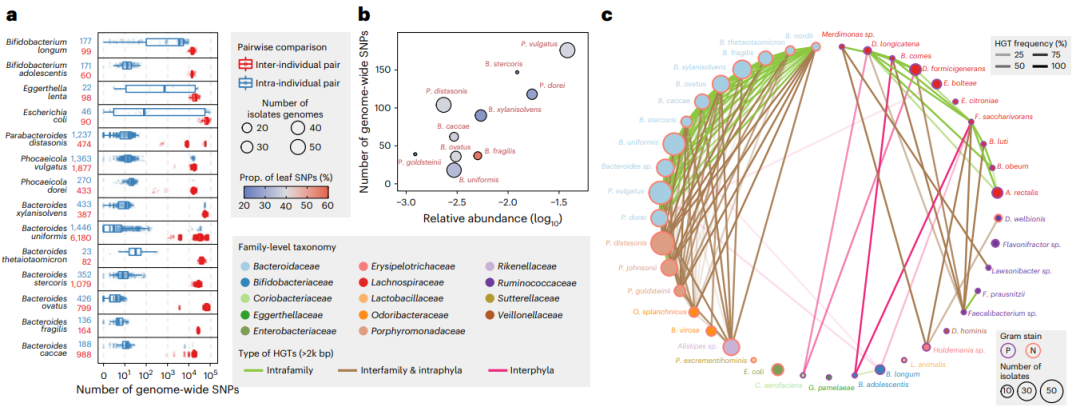
图5. 个体内和个体间肠道微生物组菌株水平的基因组多样性。a、来自同一个或不同个体的14个分离富集的菌株基因组范围的SNP数量。物种名称后面的数字代表个体间(红色)和个体内(蓝色)配对的数量。箱线图的定义:中心线:中间线;框极限:第25个四分位数上限和下限;箱须:1.5×四分位间距。b、 全基因组SNPs数量与在个体H1原始粪便样品中富含分离物物种相对丰度的相关性。点的大小表示此分析中使用的分离株的数量,颜色表示SNPs仅存在于一个基因型中的比例。c、409个H1个体中的分离株的频率2kb +HGT 的网络图。节点代表细菌种类是由科水平决定的。节点大小与分离菌株的数量成比例,边缘不透明度是两个相连物种的HGT频率。边缘颜色代表不同类型的HGT,即门间、门内和科间,或科内HGT,节点外轮廓颜色代表该物种的革兰氏染色。肠道脂质吸收来预防代谢综合征
接下来,通过分析来源于H1个体的408个菌株基因组,我们试图评估单个个体内的菌株水平多样性(补充图10;方法)。由于肠道中丰富的菌群物种被认为是经历了更多的细胞分化,因此,我们假设它们可能在基因组中积累更多的SNPs,假设肠道菌群定殖的持续时间大致相同。事实上每个分类单元中全基因组SNPs的数量通常与它在原始微生物组中的丰度相关(图5b)。B. fragilis显示出较高比例(56.0%)的叶片SNPs(即,仅存在于一个基因型),而其他物种显示出低得多的比例,包括P.goldsteini(20.5%)、B.stercoris(22.4%)和B.xylonisolvens(25.6%),这表明在物种水平上存在不同的种群瓶颈和选择性扫描。在基因水平上,我们还观察到收敛自适应进化的证据。例如,在不同的P.dorei分离株谱系,我们鉴定了在TodS基因2个编码变异(补充图12),它编码一个双组分激酶传感器,调节细菌中的甲苯代谢。甲苯和其他芳香烃存在于食品中,也被使用作为工业原料,可能污染食品,从而驱动了在肠道中的进化。
人体肠道微生物组进化的另一个主要驱动因素是水平基因转移(HGT)。因此,我们使用了H1 个体的所有分李菌株的全基因组测序数据重建了共享DNA元件的HGT网络>2 长度为kb(方法)。与与最近的报告一致,我们观察到HGT这些事件与分离株的系统发育密切相关,大多数HGT事件发生在同一门内的微生物,但在不同的科水平上甚至在不同的物种之间也普遍存在(图5c以及补充表10)。有趣的是,我们观察到HGT主要在具有相同革兰氏的分离株之间富集染色,革兰氏阴性菌表现出更普遍的HGT高于革兰氏阳性菌(P = 通过皮尔逊卡方检验为0.0005)。这一结果与最近的发现一致,表明不同的细胞壁结构可能在HGT中发挥重要作用。尤其是在我们的数据集中还观察到革兰氏阳性和阴性物种之间的HGT,启发了我们在未来研究细胞壁结构对HGT的影响并将这些HGT元素设计成微生物群落编辑工具。接下来,为了检查这些HGT是否发生,我们计算了所有物种对之间的平均HGT频率(方法)。我们假设如果HGT最近发生在两个物种之间,它们只与一小部分分离株相关,导致物种之间的频率较低。而如果HGT发生得更早并能带来增长效益,那么丰富并被后代垂直继承,导致更高频率变异。有趣的是,我们发现大多数HGT事件在分离株中存在(71.5%的HGT,频率>50%),尤其是对Bacteroidaceae物种(图5c),表明它们发生在遥远的过去,在强烈的选择下肠道环境中被富集。
考虑到个体内HGT的普遍性和高频率出现,接下来,我们注释了最广泛分布的HGT元件的蛋白质编码序列,以探索它们的潜在功能(方法)。有趣的是,我们发现了多种抗生素耐药性基因(ARGs)具有不同的作用机制和分泌系统基因(补充图13)。例如,令人惊讶的是,在 Bacteroidaceae, Porphyromonadaceae, Odoribacteraceae, and Rikenellaceae中至少13个不同的物种中,发现了前四个最广泛分布的HGT序列,包含有包括核糖体在内的多种ARG保护器和抗生素外排泵,以及III型和IV型分泌系统。而ARGs和分泌系统通过HGT可能具有明显的进化优势,在带有未知功能基因的不同物种中有许多广泛分布的元素(补充图13),暗示驱动它们在肠道中长期持久存在的未知的机制。总之,这些结果突显了这些微生物分离物在个体间和个体内具有可以使用系统表征的基因组多样性,CAMII可以基于生物库和基因组分析深入的研究个体特定的肠道微生物定殖、适应及生态学。
- 讨论 -
从肠道微生物组中分离菌株历来是以一种特殊的方式形成的,其中重要的表型特征和基因组特征一直是欠记录和描述的。这里,我们描述了CAMII平台,通过利用自动化、机器视觉、监督学习和基因组学来工业化产生菌株分离生物库。当与低成本16S测序及全基因组测序相结合,系统的生成表型和基因型数据从流水线中产生的基因组数据形成了丰富的研究微生物群落形态、多样性和进化的资源。以肠道微生物组为例,CAMII从20个健康个体中分离出大量微生物并建成菌株分离生物库,总的来说,它覆盖了所有微生物群的80%以上。该分离物集合涵盖了健康的肠道微生物,是目前最广泛的个性化分离菌株库之一。使用此资源,我们证明了菌落形态的定量分析可以预测分类学,加强了目标属的分离,并揭示了微生物之间潜在的相互作用。个体间及个体内菌株间的基因组差异的系统分析揭示了有趣的种群选择、适应和HGT模式。
这里提供的主要数据都依赖于一个共同的mGAM富集培养基,用于人类肠道微生物组的菌株分离和表征。探索替代培养基配方、其他微量营养素和宏量营养素,以及宿主或环境相关的生物化学扰动(例如,胆汁酸和外源性化合物)可以产生形态学和生长概况的变化,为肠道微生物组的未经探索的生理学和特征提供了信息。从CAMII数据集衍生出的种间相互作用关系可以是进一步用于系统地绘制微生物组动力学的驱动因素。我们设想,这些相互作用可以通过帮助识别未知的微生物衍生分子,促进了那些在本研究及其他研究中发现的顽固的“暗物质”微生物群落的生长培养。
CAMII系统使用市面上现成的组件和开源代码,这些组件和代码可以很容易地被其他人复制(见补充表1中的成分列表)。我们设想可搜索的在线门户网站将有助于共享标准化表型和基因组数据,。CAMII硬件可以进一步扩展以集成质谱测量,以获得额外的菌落特征谱,从而改进物种和代谢物的鉴定。机载自动显微镜可以进一步引入正交以微米分辨率显示微生物细胞的数据流跨越不同的频谱通道。改进的机器视觉和ML算法可以产生更好的菌株预测,并增强菌株分离的性能。
因为单个菌株是菌群复合体中的作用单元,因此,更完整的菌株收集是必须的。这种考虑了组成、种间相互作用和整个菌群代谢能力综合性的生物库,这将有助于提高研究微生物组功能、动力学和稳定性。除了人类肠道,CAMII可用于其他微物群,水生或农业环境,包括进一步的分离和分析噬菌体、真菌和原生动物。机器人自动化系统还可以有助于生成系统的菌株库,如阵列转座子插入-敲除集合或功能基因组学表达文库以及改进易处理微生物底盘的筛选用于基因工程。
参考文献
Huang, Y., Sheth, R.U., Zhao, S. et al. High-throughput microbial culturomics using automation and machine learning. Nat Biotechnol(2023).
- 通讯作者简介 -

Columbia University
Harris H. Wang
Assistant Professor
Harris Wang:an Assistant Professor at Columbia University in the Department of Systems Biology. Dr. Wang received B.S. degrees in mathematics and physics from MIT and a Ph.D. in Biophysics from Harvard.
主要研究兴趣:applying synthetic biology methods to manipulate microbial communities and mammalian systems. Using advanced approaches in genome engineering, gene synthesis, and next-generation sequencing, he studies how genomes are shaped and maintained by the environment, and how they evolve over time.
Email:hw2429@cumc.columbia.edu
猜你喜欢
iMeta简介 高引文章 高颜值绘图imageGP 网络分析iNAP
iMeta网页工具 代谢组MetOrigin 美吉云乳酸化预测DeepKla
iMeta综述 肠菌菌群 植物菌群 口腔菌群 蛋白质结构预测
10000+:菌群分析 宝宝与猫狗 梅毒狂想曲 提DNA发Nature
一文读懂:宏基因组 寄生虫益处 进化树 必备技能:提问 搜索 Endnote
16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流快速解决科研困难,我们建立了“宏基因组”讨论群,己有国内外6000+ 科研人员加入。请添加主编微信meta-genomics带你入群,务必备注“姓名-单位-研究方向-职称/年级”。高级职称请注明身份,另有海内外微生物PI群供大佬合作交流。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍未解决群内讨论,问题不私聊,帮助同行。
点击阅读原文,跳转最新文章目录阅读

















 2613
2613

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









