







冗余分析简介
什么是RDA分析?
RDA分析(Redundancy Analysis, 冗余分析),是环境因子约束化的PCA分析,可以将样本和环境因子反映在同一个二维排序图上,从图中可以直观地看出样本分布和环境因子间的关系。从概念上讲,RDA是响应变量矩阵与解释变量矩阵之间多元多重线性回归的拟合值矩阵的PCA分析,也是多响应变量回归分析的拓展。在群落分析中常使用RDA,将物种多度的变化分解为与环境变量相关的方差,用以探索群落物种组成受环境变量的约束关系。
什么是tb-RDA?
包含很多零值的物种多度数据在执行多元回归或其它基于欧氏距离的分析方法之前必须被转化,tb-RDA(基于转化的RDA)被提出用于解决这个问题。tb-RDA在分析前首先对原始数据做一定的转换,并使用转化后的数据执行RDA。即除了第一步增添了数据转化外,其余过程均和常规的RDA相同,只是在原始数据本身做了改动,RDA算法本质未变。
什么是偏冗余分析?
偏冗余分析(Partial canonical ordination, 偏RDA)相当于多元偏线性回归分析,在实际应用中同样广泛。在解释变量的向前选择过程中,偏RDA应用广泛。
什么是基于距离的冗余分析(db-RDA)?
尽管tb-RDA的应用拓展了RDA的适应范围,但无论常规的RDA或tb-RDA,样方或物种的降维过程实质上均以欧氏距离为举出。有时候我们可能期望关注非欧式距离样方或物种关系的RDA,。基于距离的冗余分析被提出用于解决这个问题,并且证明RDA能够以方差分析方式分析由用于选择的任何距离矩阵。db-RDA将主坐标分析(PCoA)计算的样方得分矩阵应用在RDA中,其好处是可以基于任意一种距离测度进行RDA排序,因此db-RDA在生态学统计分析中被广泛使用。db-RDA首先基于物种多度数据计算相异矩阵,作为PCoA的输入,之后将所有PCoA排序轴上的样方得分矩阵用于执行RDA,而不再使用原始的物种数据以及解释变量直接作为RDA的输入。由于在PCoA中可能会产生负特征值,必要时需要引入一些有效的矫正方法。尽管物种信息在相异矩阵的计算过程中丢失,但柱坐标矩阵依然可以视为表征数据总方差的距离矩阵,因此db-RDA结果反映了解释变量对从整个响应数据中得出的样方相似性的隐形。物种得分可以通过与它们所在样方得分的多度加权平均与PCoA轴建立关联而投影到最终的排序图中,用以表明响应变量对PCoA排序的贡献程度。
标签:#微生物组数据分析 #MicrobiomeStatPlot #冗余分析图 #R语言可视化 #dbRDA
作者:First draft(初稿):Defeng Bai(白德凤);Proofreading(校对):Ma Chuang(马闯) and Jiani Xun(荀佳妮);Text tutorial(文字教程):Defeng Bai(白德凤)
源代码及测试数据链接:
https://github.com/YongxinLiu/MicrobiomeStatPlot/项目中目录 3.Visualization_and_interpretation/dbRDAplot
或公众号后台回复“MicrobiomeStatPlot”领取
基于距离的冗余分析案例
这是来自于加州大学洛杉矶分校的Emeran A. Mayer团队在2023年发表于Microbiome上的一篇论文用到的dbRDA图。论文题目为:Multi-omics profles of the intestinal microbiome in irritable bowel syndrome and its bowel habit subtypes.https://doi.org/10.1186/s40168-022-01450-5
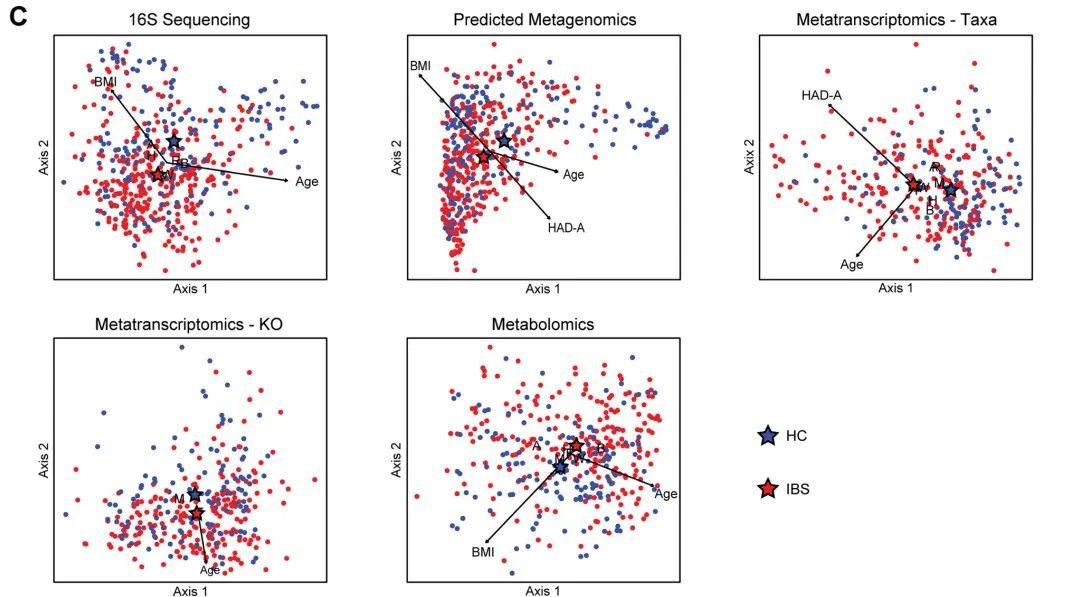
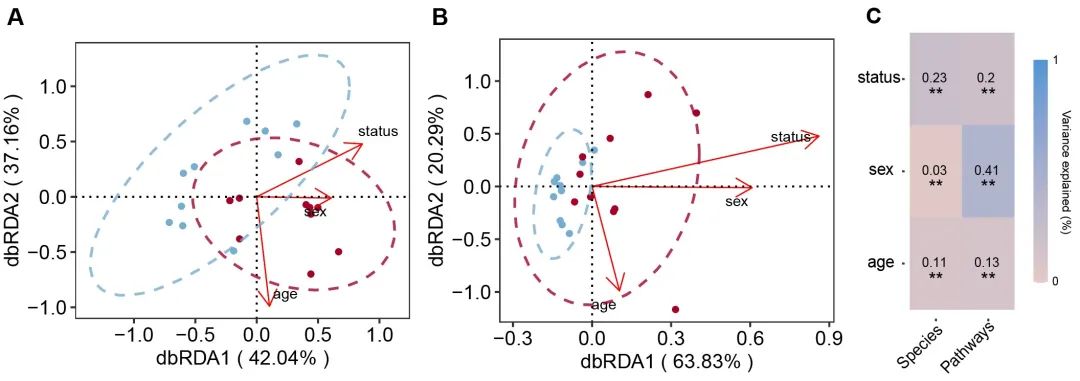
图 1 | IBS与微生物组组成和功能的全面改变有关。
图 1 (C) 进行基于距离的冗余分析 (dbRDA),以可视化与 IBS 状态、年龄、性别、种族/民族、BMI、饮食类别和 HAD-焦虑 (HAD-A) 相关的 beta 多样性变化。IBS 组和具有统计学意义的分类变量用字母或符号表示,表示每个类别的质心。具有统计学意义的连续变量显示为源自所有样本质心的箭头,长度与关联强度成正比。F = 女性,M = 男性,A = 亚裔,B = 非裔美国人,H = 西班牙裔,W = 非西班牙裔白人,R = 多种族。
结果
发现许多协变量与一个或两个数据集中的微生物组成显著相关,包括年龄、性别、种族、BMI、饮食类别、HAD-焦虑和 HAD 抑郁。鉴于焦虑和抑郁之间的强相关性以及 HAD-焦虑中 IBS 和 HC 之间的较大差异,因此将其选为协变量来表示情绪与微生物组的关联。在调整这些协变量的多变量分析中,IBS 仍然与 16S rRNA 和宏转录组测序的微生物组成显著相关,年龄和种族等几个协变量也是如此(图 1B、C)。在协变量中,年龄、性别、种族、BMI、饮食类别和 HAD-Anxiety 与宏转录组和代谢组显著相关;年龄、种族和 BMI 也与预测的宏基因组显著相关。调整这六个协变量后,IBS 仍然与宏转录组和预测的宏基因组显著相关,但与代谢组不再具有显著关联(图 1B、C)。
基于距离的冗余分析实战
源代码及测试数据链接:
https://github.com/YongxinLiu/MicrobiomeStatPlot/
或公众号后台回复“MicrobiomeStatPlot”领取
软件包安装
# 基于CRAN安装R包,检测没有则安装
p_list = c("vegan", "ggplot2", "ggpubr", "ggrepel", "rdacca.hp", "vegan", "psych", "reshape2")
for(p in p_list){if (!requireNamespace(p)){install.packages(p)}
library(p, character.only = TRUE, quietly = TRUE, warn.conflicts = FALSE)}
# 加载R包 Load the package
suppressWarnings(suppressMessages(library(vegan)))
suppressWarnings(suppressMessages(library(ggplot2)))
suppressWarnings(suppressMessages(library(ggpubr)))
suppressWarnings(suppressMessages(library(ggrepel)))
suppressWarnings(suppressMessages(library(rdacca.hp)))微生物物种dbRDA分析Microbiota species
# load data
# 读入物种数据,以细菌 OTU 水平丰度表为例
otu = read.csv('data/species_data.csv', head = T, row.names=1)
otu <- data.frame(t(otu))
# 分组数据
# group data
matadata <- read.table(paste("data/group_data.txt",sep=""), header=T, row.names=1, sep="\t", comment.char="")
otu = otu[rownames(otu) %in% rownames(matadata), ]
# 读取环境数据
# confounding factors
env = read.csv('data/c_index_species_new4.csv', header = T, row.names=1)
env = na.omit(env)
env = env[rownames(env) %in% rownames(matadata), ]
rownames = rownames(env)
rownames = as.data.frame(rownames)
otu$rownames = rownames(otu)
otu = merge(otu, rownames, by = "rownames")
rownames(otu) = otu$rownames
otu = otu[, -1]
# 根据原理一步步计算 db-RDA
# Calcula 最低0.47元/天 解锁文章
最低0.47元/天 解锁文章


















 9493
9493

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









