









文章目录
前情提要
QIIME 2 插件工作流程概述
Overview of QIIME 2 Plugin Workflows
本节内容8287字,并包括8张流程图。即是对QIIME 2工作流程的概述,也是对扩增子分析过程的高度概括和总结,建议仔细阅读,新人和老司机会有不同的感觉,但都会有很大的收获。
原文链接:https://docs.qiime2.org/2018.11/tutorials/overview/
欢迎你加入QIIME 2大家庭。本节内容将帮助你了解 QIIME 2的主要插件和可用的功能,并指导你深入学习相关的教程。换言之,本节没有回答你如何使用QIIME 2的问题,但可以指明你正确的方向。把本节当成一张藏宝图:将QIIME 2的每个功能作为你通往荣耀的垫脚石,下方的流程图将会告诉你所有的宝藏埋在哪里。
零基础上手
QIIME 2 for dummies
在我们提及插件或功能之前,对于分析扩增子数据,我们需要讨论标准QIIME 2工作流程(workflow)这一概念。在我们看概述之前,我们必须先看一下藏宝图的钥匙长什么样:

每种类型的数据(如 对象和可视化)和功能(如方法、可视化和流程)用不同颜色的节点代表。连接节点的边分为实线(代表依赖输入或输出)和虚线(代表可选输入)。没明白什么意思吗?可以回头读一下第一节中的附录。
在上面的流程图中,具体意义如下:
- 功能/动作(Actions)采用插件或功能的名称来命名。想使用这些功能,可以打在命令行,输入
qiime,再配合各种功能,如qiime demux emp-single - 流程(pipelines)是一种特殊的功能,即一条命令运行多个单独的命令。为了简洁,在一些流程图中,流程被标记为箱体 —— 封装多个在内部运行的功能。
- 对象/数据(Artifacts)采用语义类型命名的文件。别担心——除非你需要导入它们,否则你不需要记往这些长名字。
- 可视化有各种名称,一些代表数据的意义,一些用表达的意义来命名。
开始前重要的知识点
- 下面的指南并非详尽无遗。它只涵盖大多数QIIME 2“核心”插件中的一些主要操作。还有许多操作和插件需要你自己去发现和探索。如果你想学习更多?可以在终端中使用
qiime --help功能,可以列出全部功能。如具体想看dada2的功能,可以打qiime dada2 --help。如想查看拆分功能中的地球微生物组单序列拆分的帮助,打qiime demux emp-single --help。现在你学会查看每个插件和功能的方法了吗? - 下方的流程图设计的尽量简洁,因此省略了许多的输入(一些可选的特别输入和元数据)、输出(如统计摘要和次要输出)、和其它可能的功能参数。现在你学会了查阅帮助文档,可以查看并学习每个插件和功能的具体功能和参数(提示:如果有插件的功能在这里没有提到,它可能是用于检查其它功能结果的)。
- 元数据(metadata,样本信息,如分组信息)是QIIME 2中的核心概念。我们在后面有一节专门对它进行讲解和讨论。
- 对象(Artifacts
.qza)和可视化(Visualizations.qzv)文件都是标准的压缩文档,包括数据文件、分析过程可追溯的文件。可以使用unzip来解压查看内容,更好的方法是使用qiime tools export命令导出,后面的导出章节会有详细讲解。想要进一步了解这两类文件格式,可阅读数据是如何存储一文 https://dev.qiime2.org/latest/storing-data/ 。 - QIIME 2中做事情没有唯一的方法。大多数的插件和功能都是独立的软件,或现存的方法。QIIME只是起到胶水的工作,然后奇迹就发生了。通往山顶的路不只一条。
- 不要忘记合理的引用!不确定应该引用什么?查看具体功能或插件的引文,可以使用
--citations参数,在网页中查看文件也有citations的选项卡可查看。
现在我们阅读完了词汇表(第一节附录2)和金钥匙(上文),让我们全面复习一下扩增子测序数据的分析流程:
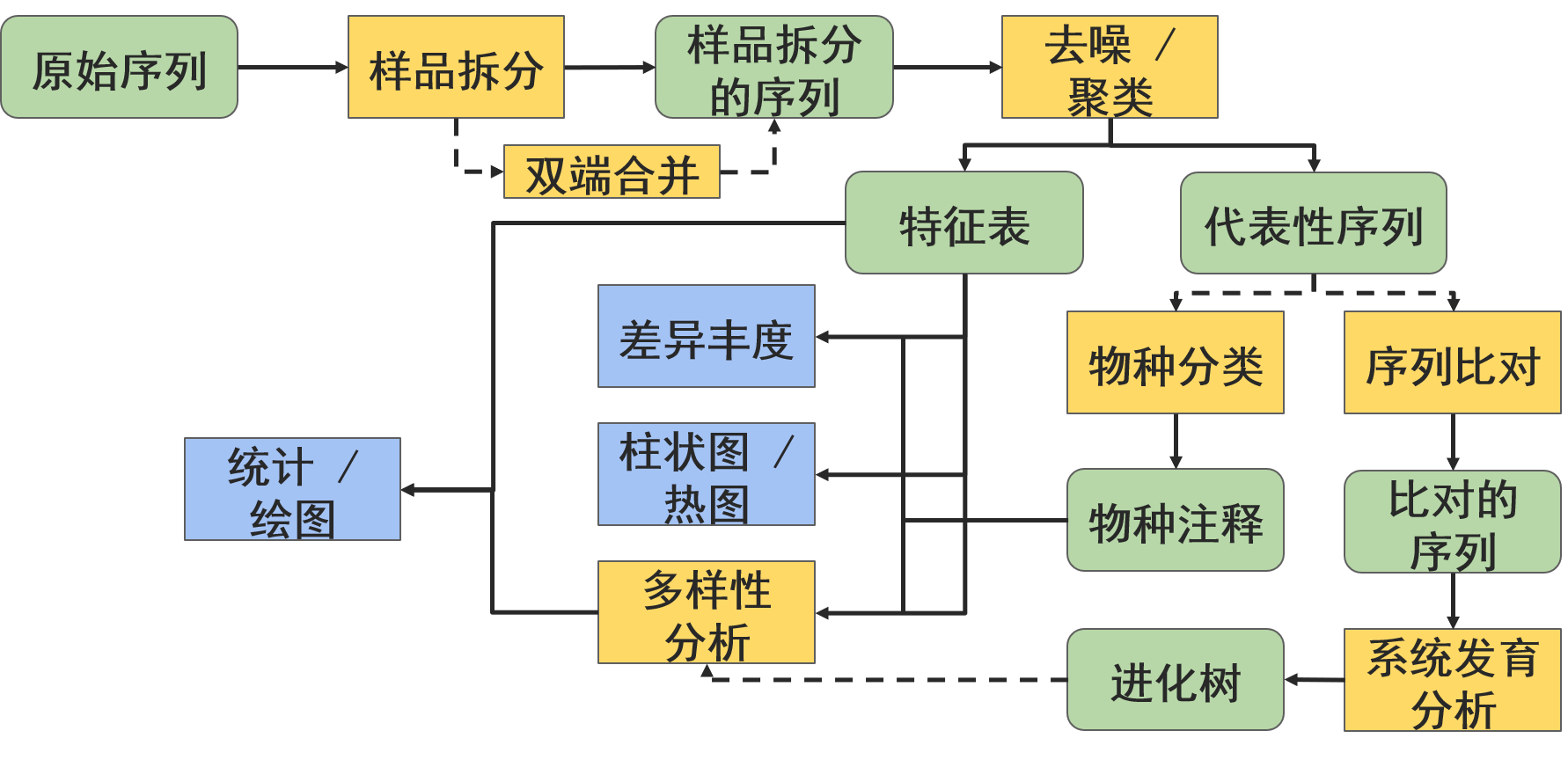
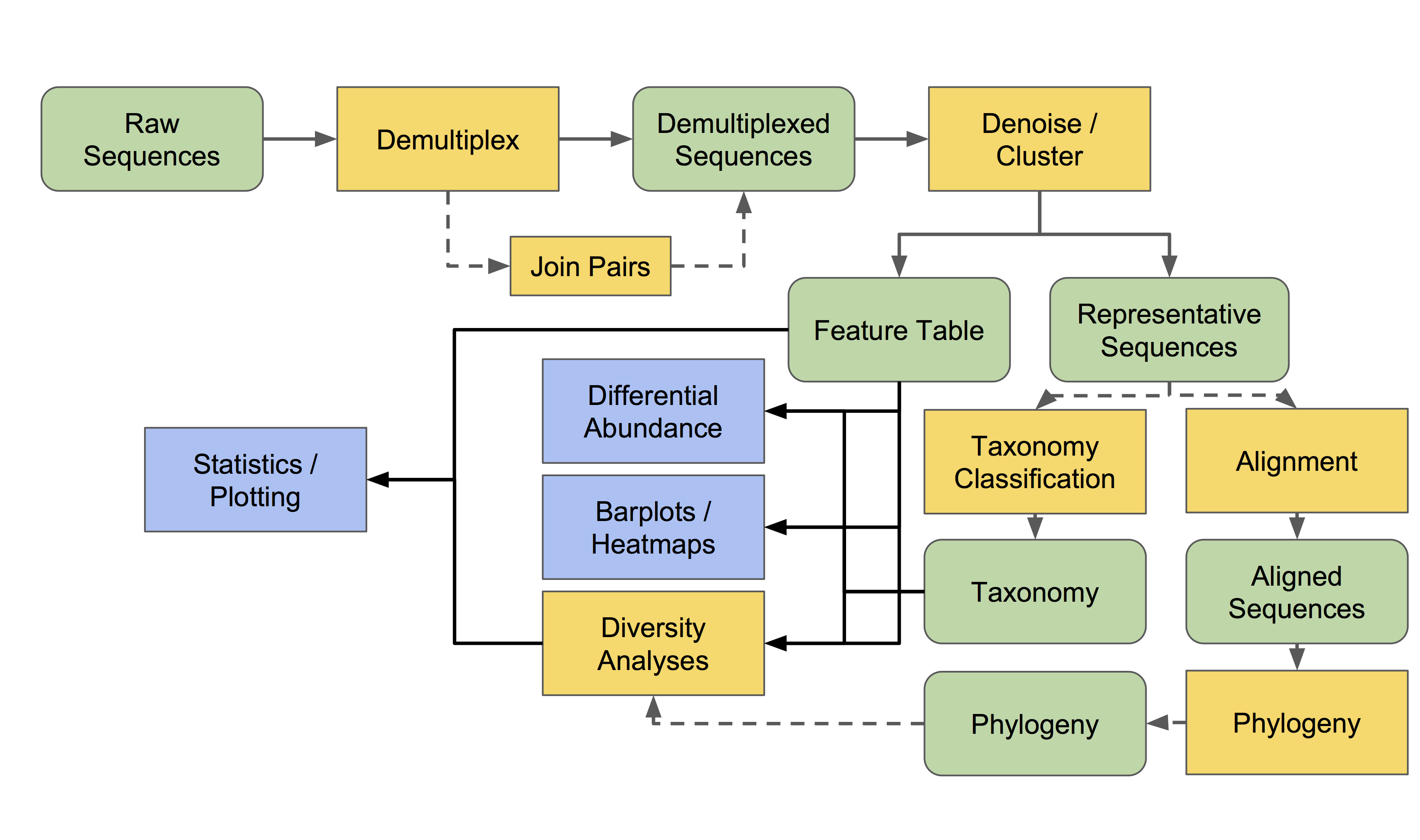
本概述中的边和节点并不表示具体的操作或数据类型,而是表示概念类别,例如,我们在实验中可能拥有的数据基本类型或分析目标。下面将更详细地讨论所有这些步骤和术语。
所有数据都必须导入为QIIME 2对象,以便由QIIME 2操作使用(除了一些元数据)。不同的用户可以在不同的阶段进入该工作流程。大多数人都有某些类型的原始序列(例如,FASTQ或FASTA)数据,这些数据应该按照适当的序列导入方案导入。其他用户可以从拆分为单样本的序列数据开始,或者从合作者给他们的特征(Features,类似于OTU但概念更广义)表开始。后面章节中的导入教程专题会详细介绍用户需要导入到QIIME 2中的最常见的数据类型。
现在我们已经了解到— — 实际上可以在几乎任何节点上进入这个工作流程,接下来让我们浏览各个部分的具体功能和应用。
- 所有的宏基因组/扩增子测序实验的分析的起点是原始序列数据。典型为fastq格式,包含有DNA序列数据和每个碱基的质量值。
- 我们必须样本拆分,确定每条序列的样本来源。
- 然后序列进行去噪(denoised)获得扩增子序列变异(amplicon sequence variants, ASVs),或聚类为可操作分类单元(operational taxonomic units, OTUs),只为以下两个目的:
- 降低测序错误;
- 序列去冗余;
- 特征(Feature,对ASV/OTU等的统称)表和代表性序列是结果的核心数据。别把它们弄丢了!一个特征表是一个样本 X 观测值的矩阵,例如:每个特征(OTUs, ASVs等)在每个样本中的观测值。
- 我们可以基于特征表做很多事,常用分析包括:
- 序列的物种分类(如,这里面有什么物种?)
- Alpha(α)和Beta(β)多样性分析,或分别描述样本内或样本间的多样性;
- 许多的多样性分析依赖于个体特征的进化相似性。如果你测序的是系统发育的标记基因,如16S rRNA基因,你可以采用多序列比对方式评估特征间的系统发育关系。
- 不同实验组间特征的差异丰度分析,确定哪些OTUs或ASVs显著的多或少。
这只是一个开始,还有众多的统计方法和绘图方法的提示,见下文。世界是你的。就让我们一探究竟吧(The world is your oyster. Let’s dive in.)?。
警告
哇! 别着急,霍斯!?我们将开始使用一些严肃的技术语言?。
还记得你在前面的部分读过的语义类型和核心概念了吗? 没有敢快返回第一章阅读,否则后果自负。⚡⚡
样本拆分 Demultiplexing
可以想象一下,我们收到一堆FASTQ数据,刚从测序仪下机还热乎的(新出锅的包子很诱人,新下机的数据你也一定有马上分析的冲动吧!)。大多数二代测序仪器有能力在单个通道(lane)/运行(run)中测序数百甚至数千个样本;我们通过多路复用/加标签混合(multiplexing)多个样本在一个文库中测序,这只是一个用来混合一整堆材料的奇特词汇。我们如何知道每个读段(Read)来自哪个样本呢?这通常采用在每个序列的一端或两端附加唯一的条形码barcode(即索引index或标签tag)序列来实现。检测这些条形码序列并将它们映射回所属的样本,允许我们对混合序列进行样本拆分。
想要开始样本拆分了吗?你(或者为你的样品建库和测序的人)应该知道哪个条形码属于哪个样品——如果你不知道,和你的实验室伙伴或测序中心谈谈。在示例元数据文件中包含此条形码信息。
样本拆分过程(如发生在QIIME 2中)看起来类似于以下工作流(现在忽略此流程图的右侧):
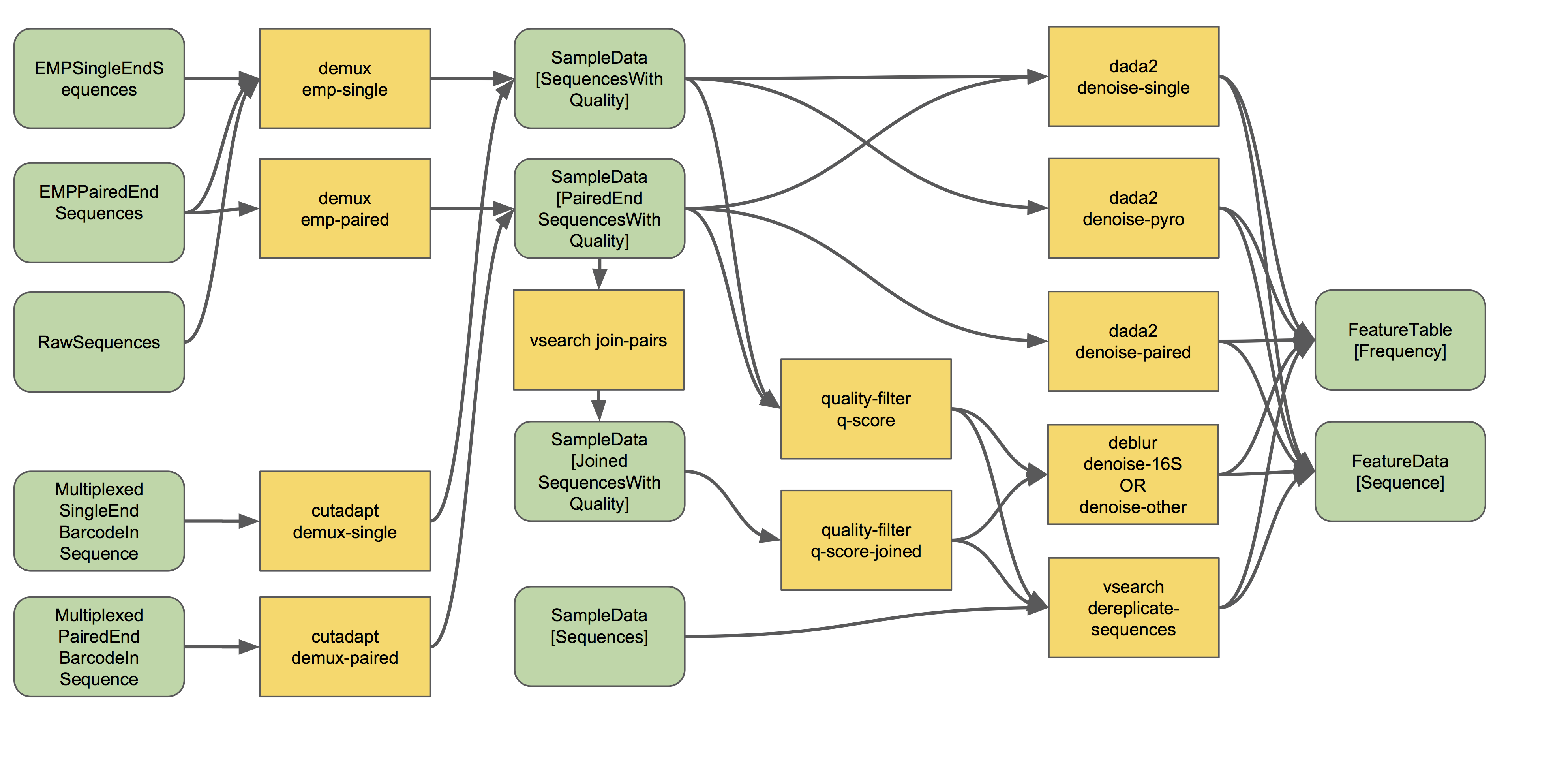
基于你导入原始数据的类型进行样本拆分,这个流程图描述了QIIME 2中当前可以处理的样本拆分类型和步骤。通常,在q2-demux或q2-cutadapt的拆分方法只有一个适用于您的数据,而这就是您所需要知道的。
阅读有关样本拆分的更多信息,并使用人体各部位微生物组教程(针对单端数据)和沙漠土壤教程(针对双端数据)对其进行拆分实战。这些教程包括地球微生物组计划(EMP)格式数据(如导入文档中所述)。你的测序数据中有条形码和引物吗?请参阅cutadapt教程以了解如何在q2-cutadapt中使用demux方法。有双端标签、无方向测序或其他不寻常的格式吗?努力祈祷?,然后查看QIIME 2论坛,看看是否有人找到解决办法(这些非标准的建库拆分方法一部分可以使用QIIME 1实现拆分和方向调整,但我强烈建议谁挖坑测序的让谁处理好了,他们奇怪的方案肯定有自己对应的个性化工具,别浪费自己的宝贵时间为别人奇怪的想法买单)。
双端测序结果在分析中某个合适的时间点,需要合并为单端。如果您按照沙漠土壤教程,您将看到在使用q2-dada2去噪期间自动进行了双端合并。但是,如果希望使用q2-debur或OTU聚类方法(见下文更详细地描述),则需要先使用q2-vsearch进行双端合并,见上方工作流程图所示。要了解有关双端序列合并的更多信息,在后面我们会有read-join的专题教程。?
如果你开始拉扯你的头发并口吐白沫,不要绝望:QIIME 2让我们在“总体概述”中学习更容易。对大多数新用户来说,导入和原始序列样本拆分数据是最头痛的部分?。但是一旦你掌握了诀窍,就很容易了。?
去噪和聚类
祝贺你走得这么远!去噪和聚类步骤比导入和样本拆分更容易! ???
这些步骤的名称非常具有描述性:
我们对序列进行去噪,用于去除和/或校正噪音序列。?
我们将序列去冗余,以减少后续步骤中的重复、文件大小和内存需求(不要担心!我们计录了每个重复的数量——read count)。?
我们对序列进行聚类,即将相似序列(例如,那些彼此≥97%的相似序列)合并为单个序列。这个过程,也称为挑选OTU,曾经是一个普遍应用的过程,用于模板去扩增的过程,最前开始流行执行一种应急的去噪过程(捕捉随机测序和PCR错误,它们应该是稀少的且与丰富的中心序列相似)。如果可以,建议使用去噪方法。世道变了,欢迎接受新时代?(与时俱进是科学的重要素质,当由于你没有跟进近几年的新方法进展,而使用了过时的方法导致了不合理的结论才是时代的悲哀)。
去噪
让我们从去噪开始,它在上图样本拆分和去噪工作流程的右边描述。
目前在QIIME 2中可用的去噪方法包括DADA2和Deblur。您可以通过阅读每个方法的原始出版物来更多地了解这些方法。DADA2的实例应用于人体各部位微生物组教程和粪菌移植教程(用于单端数据)以及阿塔卡马沙漠土壤教程(用于双端测序数据)。Deblur的示例存在人体各部位微生物组教程(针对单端数据)和双端序列合并教程(针对配对端数据)中。
注意,deblur(以及vsearch的dereplicate-seqences)分析之前必须进行数据质量过滤,但是对于dada2来说是不必要的。Deblur和DADA2都包含内部嵌合体检查方法和丰度过滤,因此按照这些方法不需要额外的过滤。 ???
简而言之,这些方法滤除有噪声的序列,校正边缘序列中的错误(仅在DADA2的分析中),去除嵌合序列,去除单体(singletons出现频率仅有一次的序列),合并去噪后的双端序列(仅在DADA2的分析中),然后对这些序列进行去冗余。
由去噪方法产生的特征有许多名称,通常是“序列变异”(sequence variant, SV)、“扩增子SV”(ASV)、“实际SV(actual SV)”、“精确SV(exact SV)”……我相信在本教程中我们已经将这些称为ASV,让我们保持术语的一致性。?
聚类
接下来我们将讨论聚类方法。去冗余(最简单的聚类方法,有效地产生100%相似度的OTU,即在数据集中观察到的所有唯一序列)也在样本拆分和去噪工作流程中描述,是QIIME 2中所有其他聚类方法的起点,如下图所示:
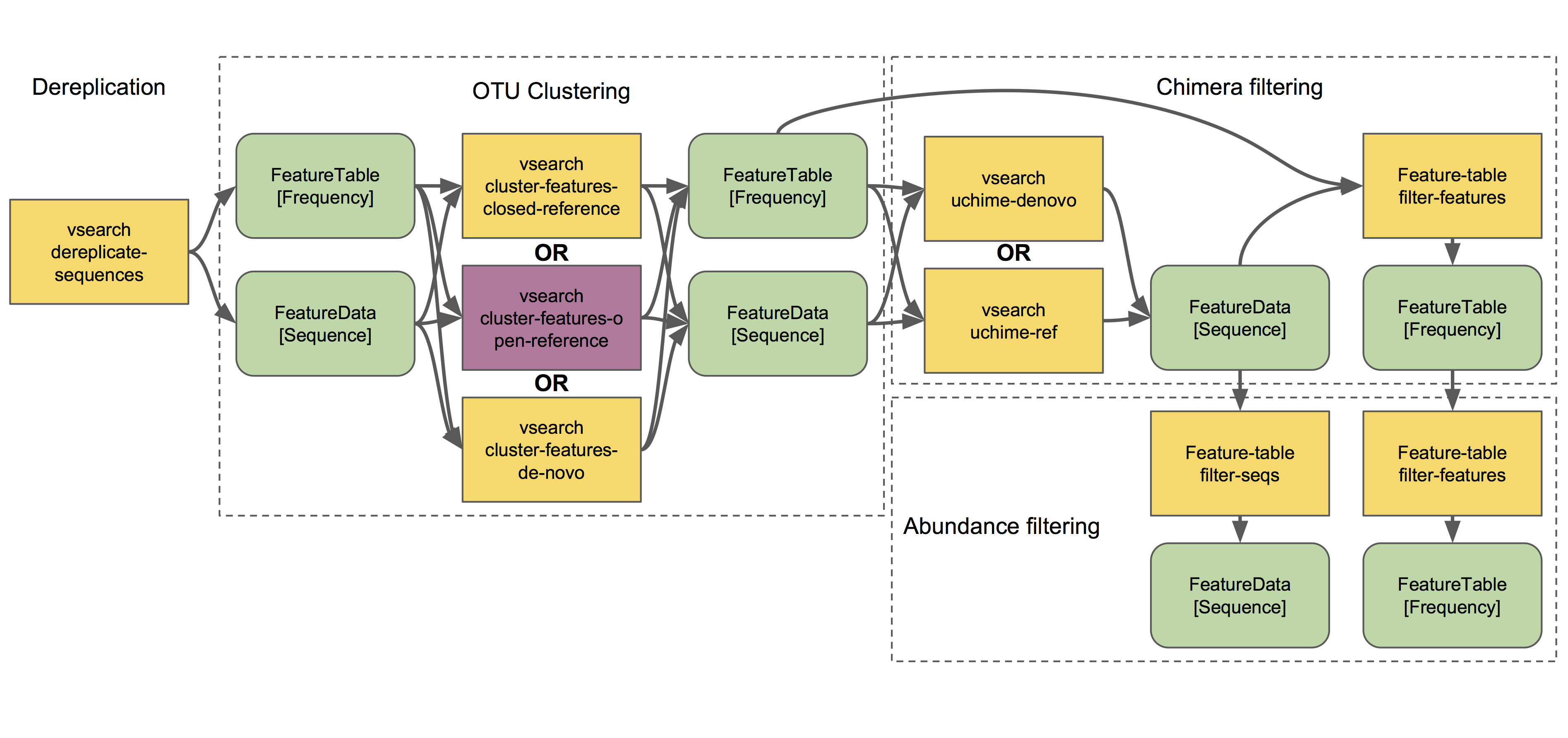
q2-vsearch实现了三种不同的OTU聚类策略:无参(de novo)、有参(closed reference)和有无参结合(open reference)。所有步骤之前都应该进行质量控制,然后进行嵌合体过滤和侵略性OTU过滤(危险的三重奏,又称Bokulich方法,可以极大的降低假阳性,但可能造成一定程度的假阴性——分析中很多时候没有最优解,只是为人的经验选择的平衡)。 ???
OTU聚类教程演示了几种q2-vsearch聚类方法的使用。别忘了阅读嵌合体过滤教程!
通过聚类方法产生的特征被称为操作分类单元(OTU),它是次优的、不精确的垃圾的世界语。 ?
译者注:在过出十年里,OTU的广泛使用为研究扩增子测序数据提供了简单快速的分析方式,带到了微生物组学文章的大爆发。但同时因其聚类的偏好性,序列可变性等缺点,使得不同研究无法可比较、可重复的问题十分严重,故被称为“次优的、不精确的垃圾的世界语——使用广泛但并不通用”
特征表
所有去噪和聚类方法/工作流的最终产品是特征表[频率Frequency](特征表feature table)对象和特征数据[序列Sequence](代表序列representative sequences)对象。这是扩增序列工作流程中最重要的两个对象,用于许多下游分析,如下面所讨论的。实际上,特征表对于任何QIIME 2分析都是至关重要的,作为每个样本的所有观察的中心记录。如此重要的对象值得拥有强大的插件q2-feature-table。我们不会在这里详细讨论这个插件的所有操作(有些在下面提到),但是它可以在特征表上执行许多有用的操作,所以要熟悉它的文档格式!?
我重复一遍:特征表是QIIME 2中分析的核心。几乎所有的分析步骤(除在样本拆分用和去噪/聚类外)都以某种方式涉及特征表。注意! ?
注意:想要查看哪个序列与特征ID相关?使用
qiime metadata tabulate命令,使用特征数据[序列Sequence]对象作为输出。
祝贺你!?您已经完成了数据的导入、样本拆分和去噪/聚类,这些对于大多数用户来说都是最复杂和困难的步骤(如果仅仅是因为有那么多方法可以做到这一点!)如果你能走这么远,剩下的就很容易了。现在开始接下来愉快的旅程。
物种分类和分类学分析
Taxonomy classification and taxonomic analyses
对于许多湿实验研究人员的目的是鉴定样品中存在的生物。例如,我的样品中有哪些属或种?这个病人的样本中有人类病原体吗?我的酒里有什么生物在游泳???
我们可以通过将要查询的序列(即我们的特征,无论是ASV还是OTU)与具有已知分类信息的参考序列数据库进行比较来获得物种注释。仅仅找到最接近的比对结果并不一定是最好的,因为其他相同或接近的序列可能具有不同的分类注释。因此,我们使用基于比对、k-mer频率等物种分类器来确定最接近的分类学关联,并具有一定程度的置信度或共识(如果不能确定地预测物种名称,那么这可能不是同一物种!)。那些对QIIME 2中的物种分类学有更多兴趣的人可以阅读,今年5月发表在Microbiome上的文章(https://doi.org/10.1186/s40168-018-0470-z),够你读到天黑(until the cows come home)。???
让我们看一下物种分类工作流的样子:
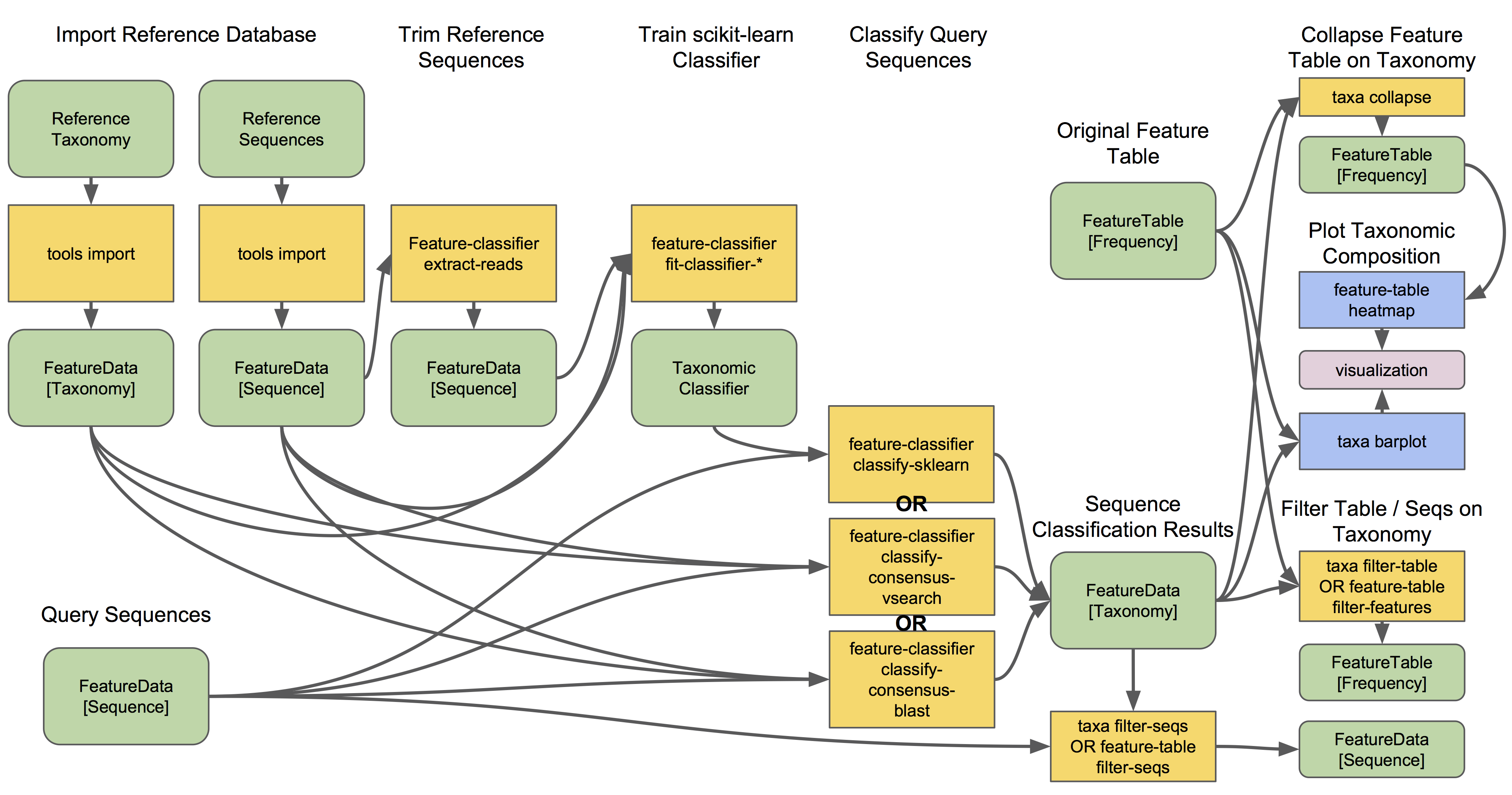
q2-feature-classifier包括三种不同的分类方法。classify-consensus-blast和classify-consensus-vsearch都是基于比对的方法,可以在N个最好的比对结果中找一致最高的用于分类。这些方法直接引用数据库FeatureData[Taxonomy]和FeatureData[Sequence]文件,不需要预先训练。
基于机器学习的分类方法是通过classify-sklearn实现的,理论上可以应用任何分类方法。必须训练这些分类器,例如,为了解哪些特征可以最好地区分每个分类学组,在分类过程中添加额外的步骤。分类器训练过程是参考数据库、特异的标记基因和且每个标记基因/参考数据库组合计算一次;然后该分类器可以多次使用而不需要重新训练!
大多数用户甚至不需要按照该教程执行训练步骤,因为可爱的QIIME 2开发人员提供了几个预先训练的分类器供大家使用。??????
哪种方法最好?它们都很好,否则我们就不麻烦把它们呈现给用户了。? 但是,一般来说,使用Naive Bayes分类器的classify-sklearn可以稍微优于我们基于16S rRNA基因和真菌ITS序列分类的几个标准测试的其他方法。然而,对于一些用户来说,这可能更加困难和令人沮丧,因为它需要额外的训练步骤。这个训练步骤可能需要大量的内存,对于某些无法使用预先训练分类器的用户来说,这将成为一个障碍。一些用户还喜欢基于比对的方法,因为这种操作模式更加透明并且参数更易于操作 (参见后面会讲到的数据 资源章节,以了解这些参数的描述以及不同应用的推荐设置)。
特征分类过程可能会比较慢。这完全取决于你序列的数量和参考序列的数量。与进行分类相比,OTU序列聚类需要更长的时间(因为序列通常更多)。如果担心运行时间太长,在分类之前从序列文件中筛选排除低丰度的特征,并可以使用较小的参考数据库。实际上,在“正常大小”的测序实验中(无论这意味着什么?),我们需要消耗几分钟(几百个特征)到几个小时(几十万个特征)之间来完成分类。如果您希望在看到精确的时间数字,请查看我们的分类器运行时性能评估和测试的结果。?⏱️
特征分类可以是内存密集型的。我们通常看到最小4GB的RAM,最大32GB的要求。这完全取决于参考序列的大小、长度和特征序列的数量……
使用classify-sklearning的示例在特征分类器教程和人体各部微生物组教程中出现。分类的流程图使其他分类器方法更加清晰。
所有分类器生成一个FeatureData[Taxonomy]对象,其中包含每个查询序列的物种分类信息。
提示: 想要查看哪个序列和分类学与特征ID相关?使用
qiime metadata tabulate命令,使用特征数据FeatureData[序列Sequence]和FeatureData[Taxonomy]对象作为输出。
序列分类注释后
物种分类是打开新世界成为可能 ?
我们现在拥有了特征数据 FeatureData[物种注释Taxonomy] 对象:
- 按分类层次相同的分类单元进行合并!这将共享相同分类的所有特征合并为单个特征。该分类单元名称成为新特征表中的特征ID。这个特征表可以与原始表相同的方式使用。一些用户可能对这类表特别感兴趣,例如,可以进行基于不同分类层级的多样性分析,但大家都对这些分类群的差异丰度分析更感兴趣。比较使用分类群作为特征的差异丰度分析,与使用ASV或OTU作为特征的差异丰度分析都可以为各种分析提供诊断和信息。 ?
- 绘制物种组成,方便查看每个样本中各种分类群的丰度。查看物种丰度条形图和特征表热图了解更多细节。?
- 过滤特征表和代表序列(
FeatureData[Sequence]对象)以删除某些分类学组。这对于去除已知的污染物或非目标基团是有用的,例如,宿主DNA,包括线粒体或叶绿体序列。对于集中于特定组以进行更深入的分析,也是有用的。有关更多细节和示例,请参阅过滤教程。??
多序列比对和进化树构建
Sequence alignment and phylogeny building
许多多样性分析依赖于个体特征之间的系统发育相似性。如果你正在测序系统发育标记(例如,16S rRNA基因),你可以多序列比对/对齐这些序列来评估每个特征之间的系统发育关系。然后这个系统发育树可以被其他下游分析使用,例如UniFrac距离分析。
用于对齐序列和产生系统发育的不同方法展示在下面的流程图中。有关多序列比对/系统发育构建的详细描述,请参阅q2-phylogeny和q2-fragment-insertion教程。 ?

现在我们拥有了系统发育树Phylogeny[Rooted有根系] 对象,请注意在接下来的分析中哪些地方使用到它 ?
多样性分析
Diversity analysis
在微生物学实验中,研究人员经常对以下事情感到困惑:
- 我的样本中有多少不同的物种/OTU/ASV?
- 每个样本存在多少系统发育多样性?
- 单个样本和样本组有多相似/不同?
- 哪些因素(仅举几个例子,如pH、海拔、血压、身体部位或宿主物种)与微生物组成和多样性的差异相关?
还有更多。这些问题可以通过α和β多样性分析来回答。Alpha多样性测量单个样本中的多样性水平。β多样性测量样本之间的多样性或差异程度。然后我们可以使用统计检验来说明样本组之间的α多样性是否不同(例如,指出哪些组具有更多/更少的物种丰富度)以及组之间的β多样性是否显著差异(例如,确定一个组中的样本比另一个组中的样本更相似),通过这些结果来证明这些组中的成员正在形成一个特定的微生物组成。
QIIME 2中不同类型的多样性分析在人体各部微生物组教程和粪菌移植教程中进行演示,这里将演示用于生成多样性对象的全套分析(并非全部:注意,其他插件可以在这些对象上进一步分析,本指南中也将进一步说明):
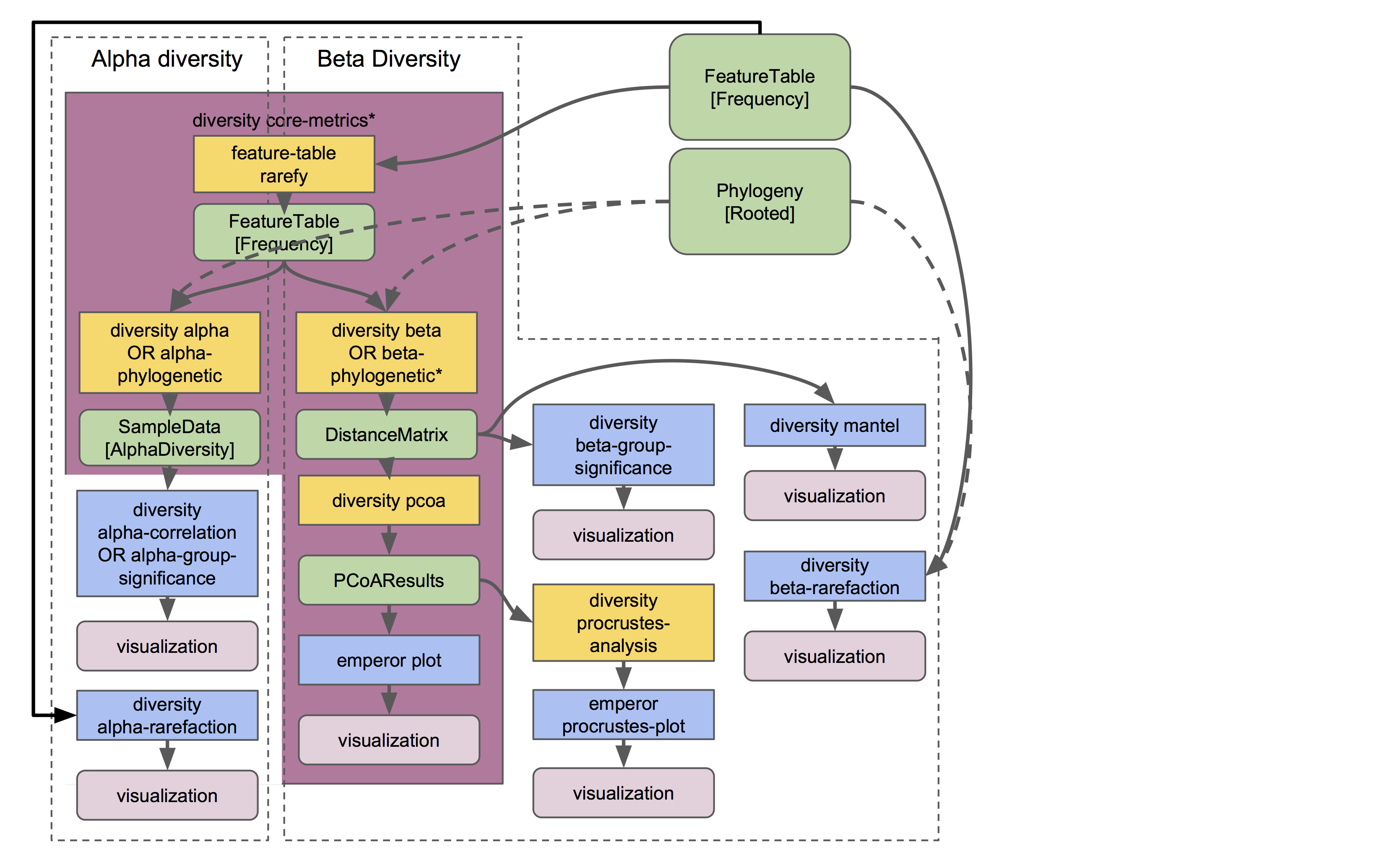
q2-diversity插件包含许多不同的有用操作!看看他们可以干什么很重要。正如您在流程图中看到的,diversity core-metrics*流程(core-metrics 和 core-metrics-phylogenetic)包含许多不同的核心多样性命令,并且在这个过程中产生可用于下游分析的主要对象。它们是:
SampleData[AlphaDiversity]对象,其中包含特征表中每个样本的α多样性估计。这是α多样性分析的核心对象。DistanceMatrix对象,包含特征表中每对样本之间的成对距离/差异。这是β多样性分析的核心对象。PCoAResults对象,包含每个距离/不同度量的主坐标排序结果。主坐标分析是一种降维技术,有助于在二维或三维空间中进行样本相似度可视化比较。
这些是与多样性相关的主要对象。记住它们的名字和保存位置!我们将在各种下游分析中,或在流程图中所示的q2-diversity的各种操作中用到这些数据。这些功能大部分操作会在人体各部位微生物组教程中演示,所以请继续加油学习更多姿势吧!☔
注意,在QIIME 2中有许多不同的α和β多样性度量方法。要了解更多(并找出你应该引用谁的论文!),请阅读由QIIME 2用户整理的帖子 https://forum.qiime2.org/t/alpha-and-beta-diversity-explanations-and-commands/2282/3 ,适合我们所有人查阅。感谢斯蒂芬妮的辛苦总结整理! ??????
玩转特征表
Fun with feature tables
此时,您有一个特征表、物种分类结果、α多样性和β多样性结果。哦,我的天哪! ?
如上所述,物种和多样性分析是大多数QIIME 2用户需要执行的基本分析类型。然而,这仅仅是分析的开始,还有很多更高级的分析等待我们去挖掘和探索?️⌨️
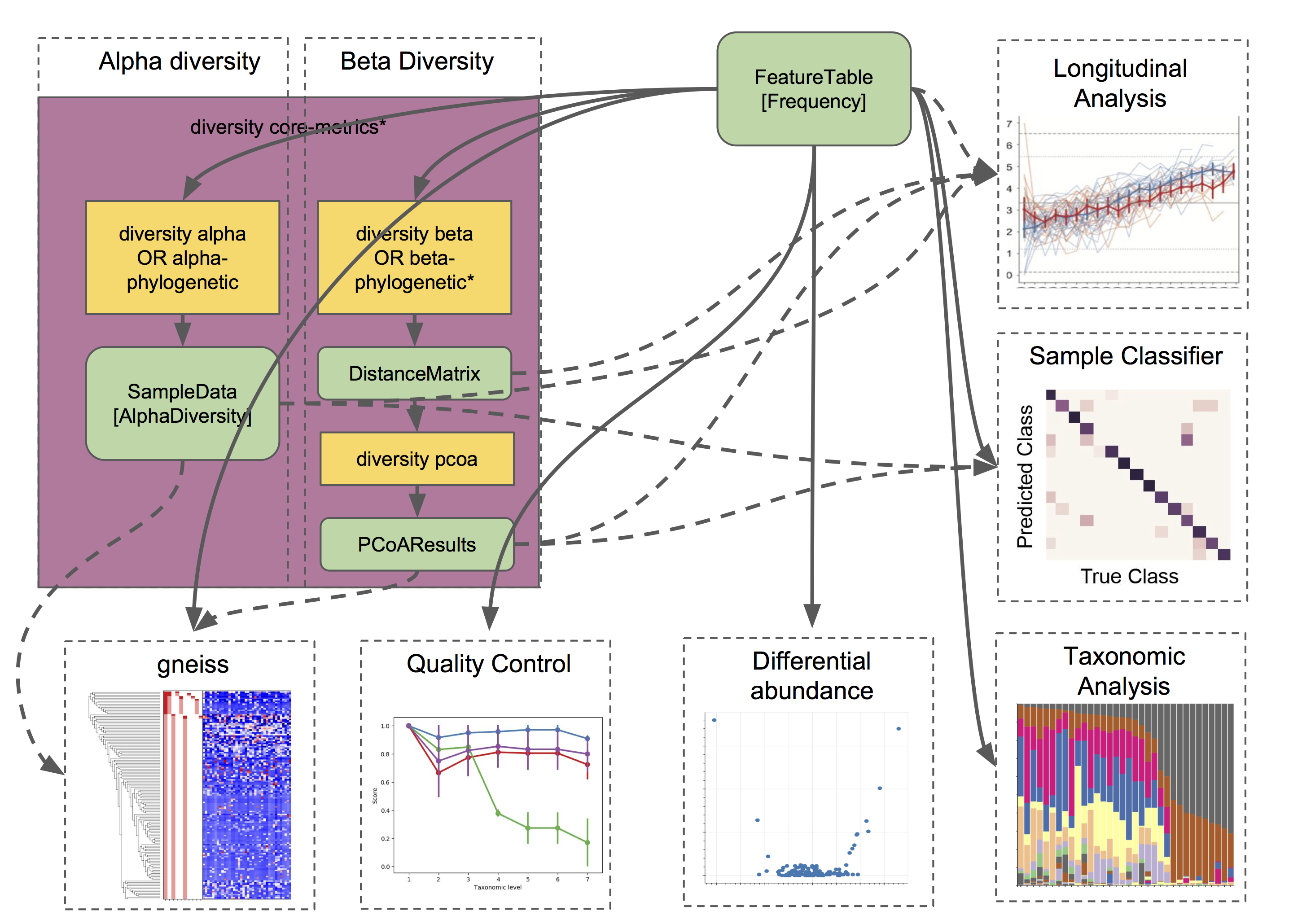
我们这里只是给出一个简要的概述,因为每种分析都有自己详细的教程来指导我们:
- 分析纵向数据:
q2-longitudinal是一个用于执行纵向实验的统计分析插件,也就是说,样本是随着时间重复地从单个患者/受试者/站点收集的。这包括对α和β多样性的纵向研究,以及一些非常棒的交互式图形。?? - 预测未来(或过去) ?:
q2-sample-classifier是用于特征数据机器学习? 分析的插件。支持分类和回归模型。这允许您执行以下操作:- 根据特征数据预测样本元数据(例如,我们能否使用粪便样本预测癌症易感性?或者根据发酵前葡萄的微生物组成预测葡萄酒质量??
- 识别预测不同样本的特征。?
- 定量微生物组发育阶段(例如,跟踪婴儿肠道中正常的微生物组发育受持续营养不良、抗生素、饮食和分娩方式的影响)。?
- 预测异常值和标签错误的样本。?
- 差异丰度用于确定在不同样本组中哪些特征显著更多/更少。QIIME 2目前支持几种不同的差异丰度统计检验方法,包括ANCOM(
q2-composition中的一个功能)和q2-gneiss??? - 评价和控制数据质量:
q2-quality-control是评价和控制序列数据质量的插件。它包括以下功能:- 测试不同生物信息学或分子方法的准确性,或批次(run-to-run)的变异。常用于研究组成已知的样本,如人工重组群体(mock communities,模拟群体,即人为按一定比例混合多种微生物的重组群体),可以研究观察值和预期组成、序列等之间的相似性,来评估实验、分析方法对结果的影响等。以及更多有创造力的实验和应用…?
- 基于对参考数据库或包含特定DNA短片段(例如,引物序列)比对,进行序列的过滤。这对于去除特定生物组、非目标DNA或其他没有意义的序列是非常有效的。?
这只是一个简单的概述!QIIME 2正在持续增长,因此请继续关注未来版本中的更多插件?,并关注继续扩展QIIME 2中可用的第三方插件。?
现在开始享受分析的愉悦之旅吧!?
译者简介

刘永鑫,博士。2008年毕业于东北农大微生物学专业。2014年中科院遗传发育所获生物信息学博士学位,2016年博士后出站留所工作,任宏基因组学实验室工程师,目前主要研究方向为宏基因组学、数据分析与可重复计算和植物微生物组、QIIME 2项目参与人。发于论文12篇,SCI收录9篇。2017年7月创办“宏基因组”公众号,目前分享宏基因组、扩增子原创文章300+篇,代表博文有《扩增子图表解读、分析流程和统计绘图三部曲》,关注人数3万+,累计阅读400万+。
Reference
- https://qiime2.org/
- Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet C, Al-Ghalith GA, Alexander H, Alm EJ, Arumugam M, Asnicar F, Bai Y, Bisanz JE, Bittinger K, Brejnrod A, Brislawn CJ, Brown CT, Callahan BJ, Caraballo-Rodríguez AM, Chase J, Cope E, Da Silva R, Dorrestein PC, Douglas GM, Durall DM, Duvallet C, Edwardson CF, Ernst M, Estaki M, Fouquier J, Gauglitz JM, Gibson DL, Gonzalez A, Gorlick K, Guo J, Hillmann B, Holmes S, Holste H, Huttenhower C, Huttley G, Janssen S, Jarmusch AK, Jiang L, Kaehler B, Kang KB, Keefe CR, Keim P, Kelley ST, Knights D, Koester I, Kosciolek T, Kreps J, Langille MG, Lee J, Ley R, Liu Y, Loftfield E, Lozupone C, Maher M, Marotz C, Martin BD, McDonald D, McIver LJ, Melnik AV, Metcalf JL, Morgan SC, Morton J, Naimey AT, Navas-Molina JA, Nothias LF, Orchanian SB, Pearson T, Peoples SL, Petras D, Preuss ML, Pruesse E, Rasmussen LB, Rivers A, Robeson, II MS, Rosenthal P, Segata N, Shaffer M, Shiffer A, Sinha R, Song SJ, Spear JR, Swafford AD, Thompson LR, Torres PJ, Trinh P, Tripathi A, Turnbaugh PJ, Ul-Hasan S, van der Hooft JJ, Vargas F, Vázquez-Baeza Y, Vogtmann E, von Hippel M, Walters W, Wan Y, Wang M, Warren J, Weber KC, Williamson CH, Willis AD, Xu ZZ, Zaneveld JR, Zhang Y, Zhu Q, Knight R, Caporaso JG. 2018. QIIME 2: Reproducible, interactive, scalable, and extensible microbiome data science. PeerJ Preprints 6:e27295v2 https://doi.org/10.7287/peerj.preprints.27295v2
猜你喜欢
- 10000+: 菌群分析
宝宝与猫狗 提DNA发Nature 实验分析谁对结果影响大 Cell微生物专刊 肠道指挥大脑 - 系列教程:微生物组入门 Biostar 微生物组 宏基因组
- 专业技能:生信宝典 学术图表 高分文章 不可或缺的人
- 一文读懂:宏基因组 寄生虫益处 进化树
- 必备技能:提问 搜索 Endnote
- 文献阅读 热心肠 SemanticScholar Geenmedical
- 扩增子分析:图表解读 分析流程 统计绘图
- 16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
- 在线工具:16S预测培养基 生信绘图
- 科研经验:云笔记 云协作 公众号
- 编程模板: Shell R Perl
- 生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流、快速解决科研困难,我们建立了“宏基因组”专业讨论群,目前己有国内外2600+ 一线科研人员加入。参与讨论,获得专业解答,欢迎分享此文至朋友圈,并扫码加主编好友带你入群,务必备注“姓名-单位-研究方向-职称/年级”。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍末解决群内讨论,问题不私聊,帮助同行。

学习扩增子、宏基因组科研思路和分析实战,关注“宏基因组”


点击阅读原文,跳转最新文章目录阅读
https://mp.weixin.qq.com/s/5jQspEvH5_4Xmart22gjMA














 6713
6713











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









