







 本文详细介绍了单细胞转录组分析的步骤,包括数据预处理、归一化、降维(PCA)、聚类和细胞分类。通过PCA和UMAP进行数据可视化,并利用louvain算法进行细胞聚类。同时,确定了高变基因并进行了标记基因的检索,最终将细胞分为兴奋神经元、抑制神经元和非神经元三大类。
本文详细介绍了单细胞转录组分析的步骤,包括数据预处理、归一化、降维(PCA)、聚类和细胞分类。通过PCA和UMAP进行数据可视化,并利用louvain算法进行细胞聚类。同时,确定了高变基因并进行了标记基因的检索,最终将细胞分为兴奋神经元、抑制神经元和非神经元三大类。
本文将详细讲述单细胞转录组分析的步骤。
一、文章信息
题目:ClusterMap for multi-scale clustering analysis of spatial gene expression
链接:https://doi.org/10.1038/s41467-021-26044-x
期刊:Nature
二、数据集
数据集:小鼠初级视觉皮层V1_1020
转录组技术:STARmap
基因数:1020
细胞数:1650
总表达数:471295
细胞类型:16种
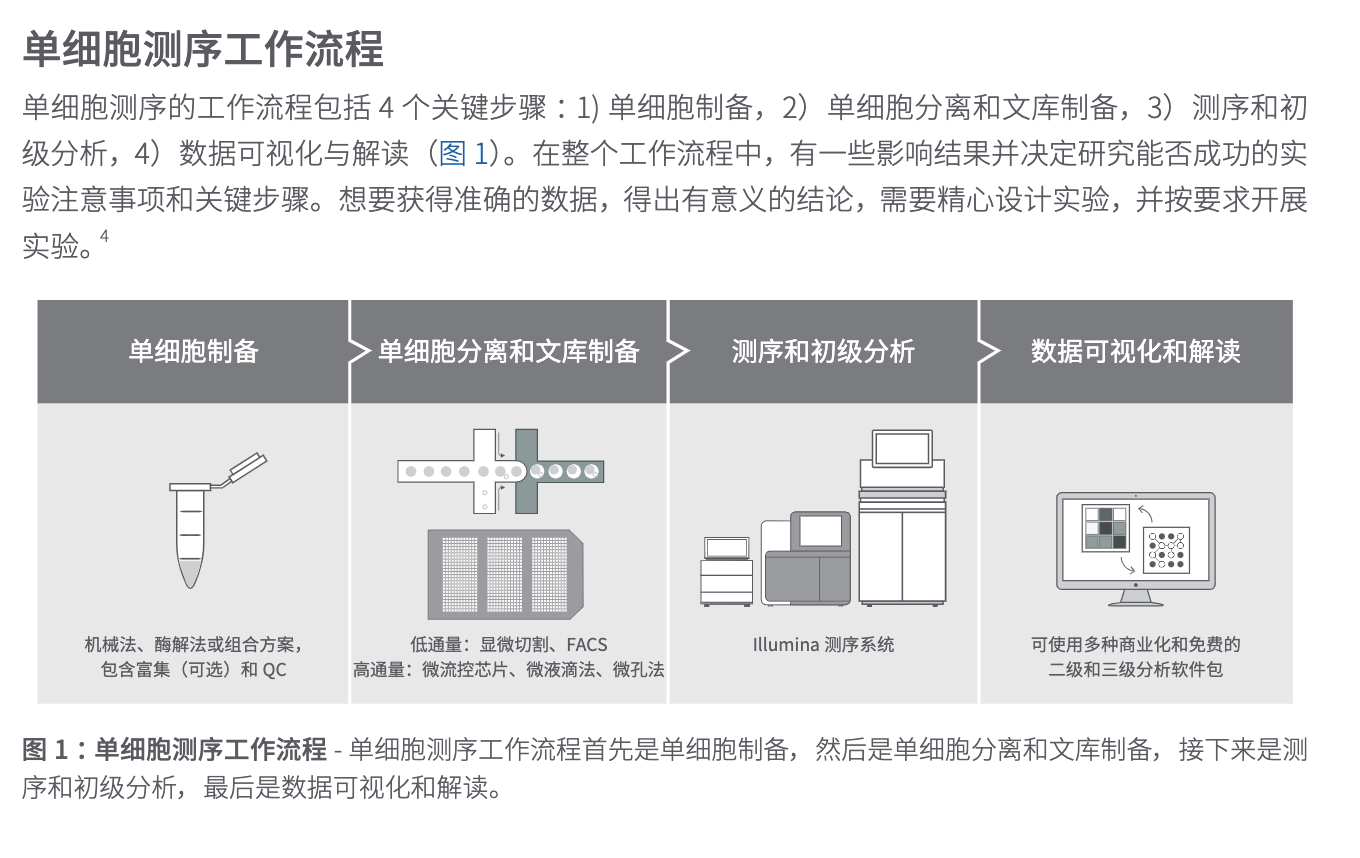
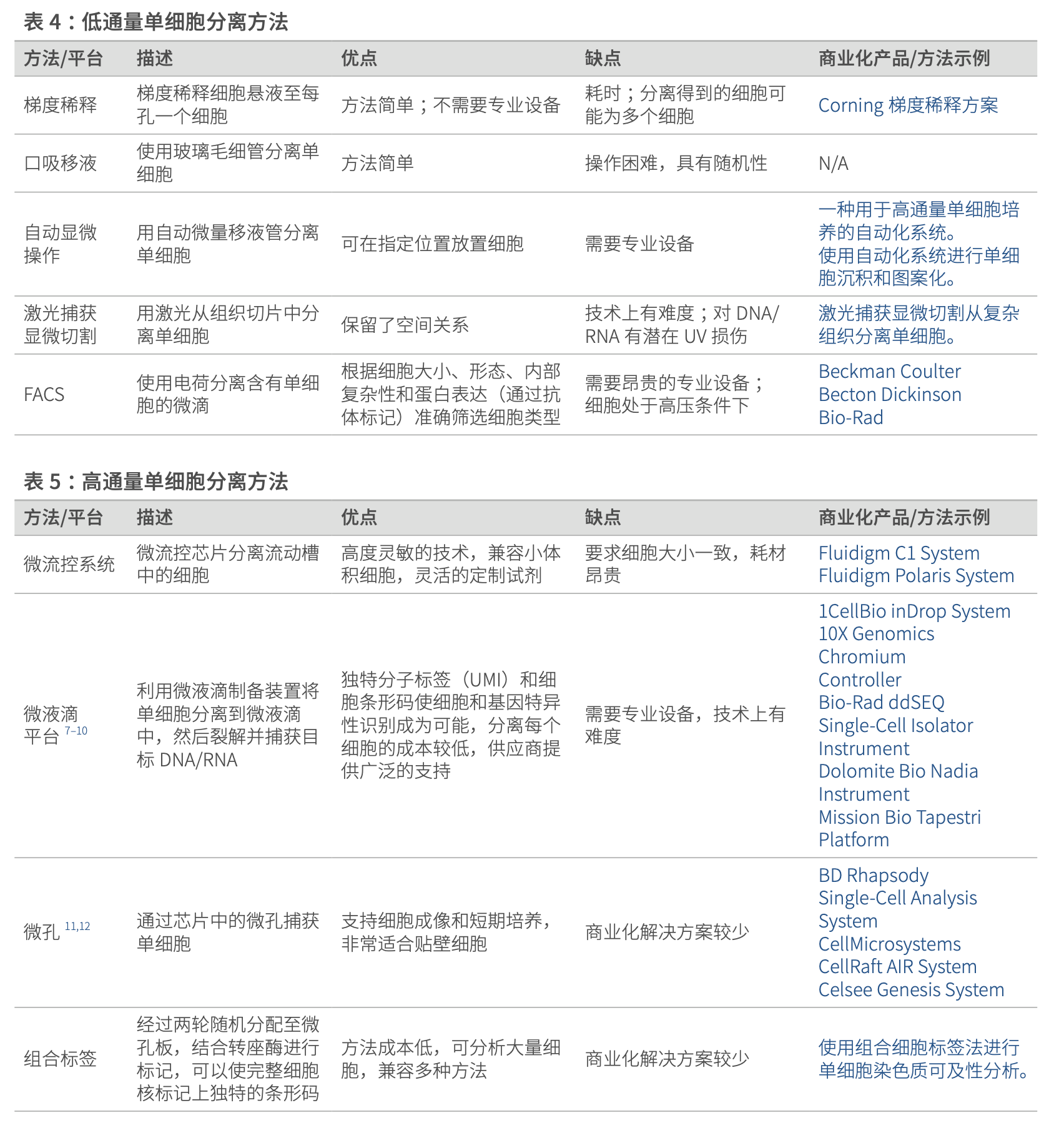
三、单细胞测序工作
关于这一部分,这篇文章没有讲解,我在了解生物背景的时候,查阅了一些资料,关于单细胞最初的制备工作。
对于这篇文章,在作者给的公开代码和数据中,数据已经是处理好的有关所有基因点的三维位置坐标。
四、细胞分类
进行细胞分类的关键是得到有细胞位置的数据集。
1、输入文件
文件由三部分组成,X,obs,var。三者的位置关系如下图所示:
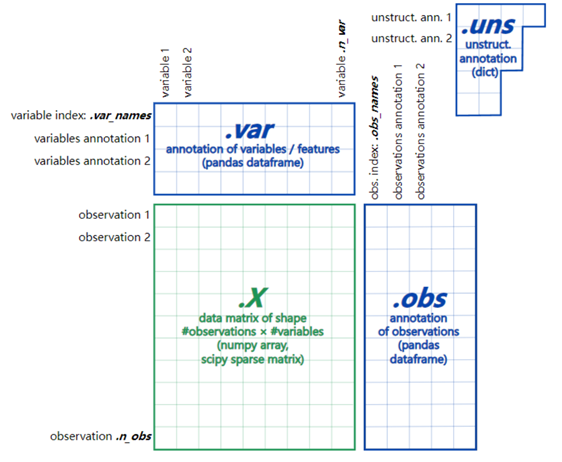
输入文件,得到adata数据格式,但是下文的操作都在细胞层面,所以对应查看adata.obs文件。
expr_path = os.path.join(out_path, 'expr_BY1.csv') # 里面全是0 1 2 3 的数字
var_path = os.path.join(out_path, 'var_BY1.csv') # 基因的名称
obs_path = os.path.join(out_path, 'obs_BY1.csv') # 二维行列坐标
# add expression data to the AnnData object 添加表达式数据到AnnData对象
expr_x = np.loadtxt(expr_path, delimiter=',') # delimiter表示分隔符,加载文件的分隔符
var = pd.read_csv(var_path, header=None) #header=None(文件中不包含列名的行)
var = pd.DataFrame(index=var.iloc[:,0].to_list())
obs = pd.read_csv(obs_path, index_col=0)
adata = AnnData(X=expr_x, var=var, obs=obs)2、预处理
(1)可视化高表达基因
sc.pl.highest_expr_genes(adata, n_top=20) 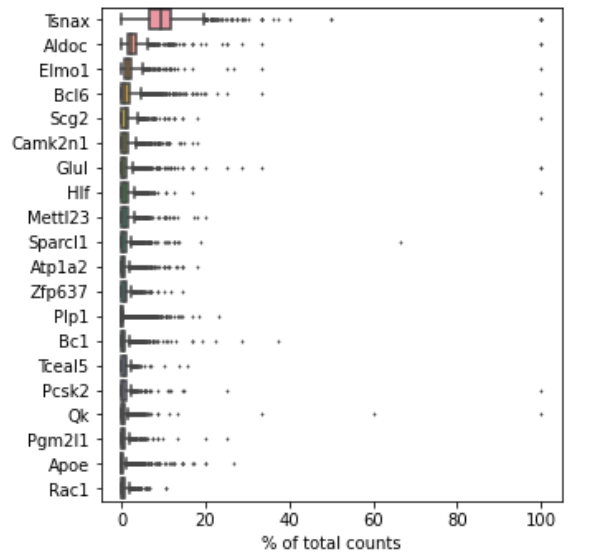
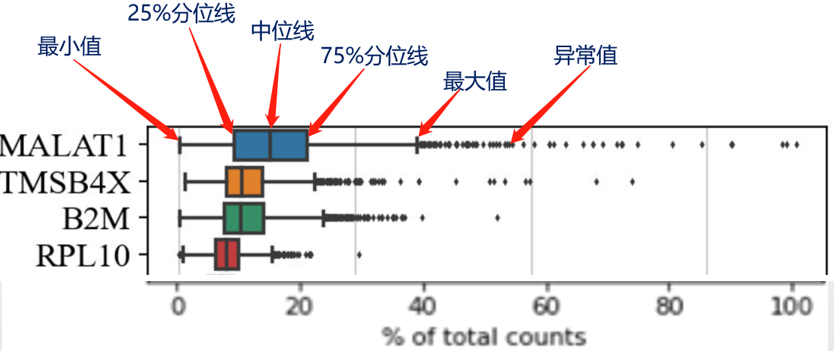
可视化所有细胞中表达量最多的20个基因,展示的方法采用的是箱线图。箱线图是对每个基因在所有细胞中表达量分布的更详细描述。箱子的宽度,反应了数据的波动程度,箱子越窄,数据分布越集中。
箱线图的查看以下图为例:中位线表示的是MALAT1在单个细胞表达量的中位数;中位数对应的是该基因表达量占该细胞总基因表达量的17%。可见,MALAT1基因在大部分细胞中的表达量占比约在10%~20%之间。
 最低0.47元/天 解锁文章
最低0.47元/天 解锁文章

















 2212
2212

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









