







1. 软件下载
https://choishingwan.github.io/PRS-Tutorial/prsice/
最新版的包括Mac和Linux系统,下面我们用Linux系统进行演示
安装包:
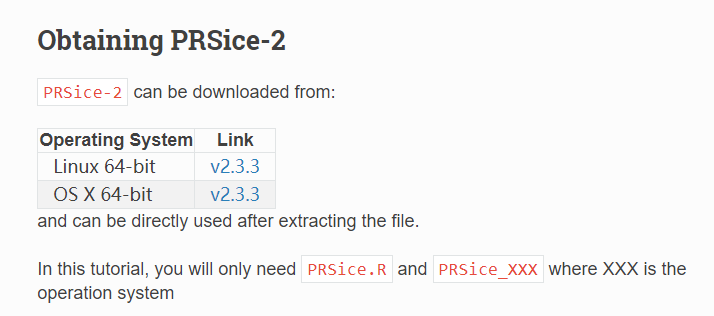
测试数据:
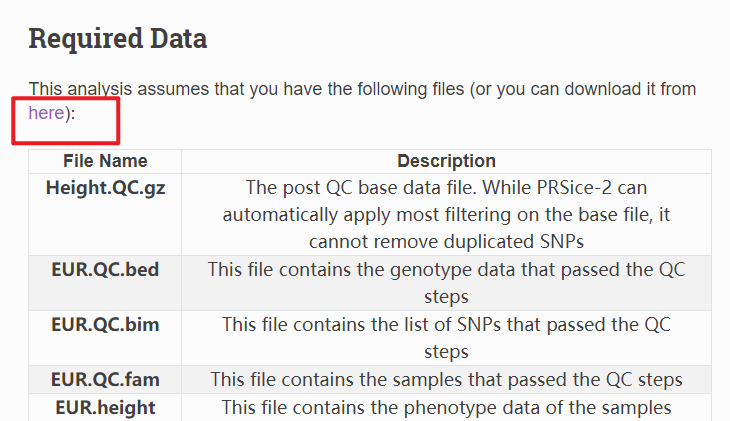
注意,上面数据如果无法下载,可以公众号(育种数据分析之放飞自我)后台回复PRS,获得软件包和测试数据。
2. 软件安装
本演示,在Linux系统下Centos7进行。
下载好的安装包和测试数据:
新建一个prs文件夹,将压缩包放进去,解压软件:
$ unzip PRSice_linux.zip
Archive: PRSice_linux.zip
inflating: PRSice.R
inflating: TOY_BASE_GWAS.assoc
inflating: TOY_TARGET_DATA.bed
inflating: TOY_TARGET_DATA.bim
inflating: TOY_TARGET_DATA.fam
inflating: TOY_TARGET_DATA.pheno
inflating: PRSice_linux
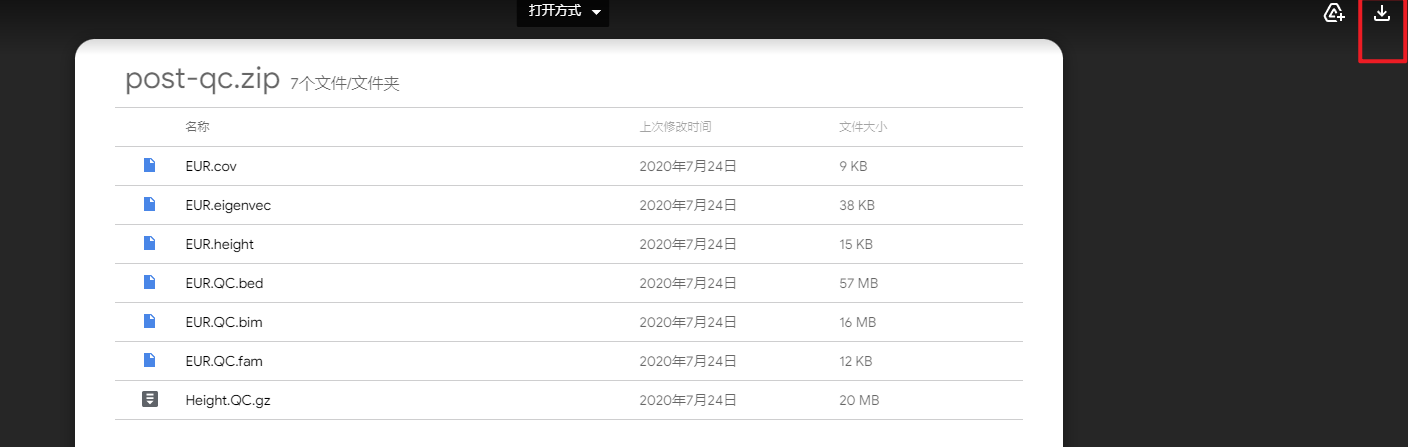
新建一个压缩包,把测试数据放进去,解压:
$ unzip post-qc.zip
Archive: post-qc.zip
inflating: Height.QC.gz
inflating: EUR.QC.bed
inflating: EUR.QC.bim
inflating: EUR.QC.fam
inflating: EUR.height
inflating: EUR.cov
inflating: EUR.eigenvec
3. 命令文件介绍
3.1 PRSice_linux
二进制文件,直接运行即可。核心计算文件,计算PRS。
3.2 PRSice.R
R 脚本文件,主要是绘图可视化,包括bar plot, high-resolution plot和quantile plot
3.3 BESE文件
基础数据文件,这里是GWAS summary的结果。
包括91063个snp结果。
TOY_BASE_GWAS.assoc
数据包括:
- SNP名称
- 染色体
- 物理位置
- A1,有效的分型,effective allel
- A2,无效的分型,non-effective allel
- P,P值
- OR,OR值
$ head TOY_BASE_GWAS.assoc
SNP CHR BP A1 A2 P OR
SNP_22857 4 103593179 1 2 0.2852 13.29
SNP_13879 2 237416793 1 2 0.8784 21.624
SNP_20771 4 16957461 1 2 0.1994 91.265
SNP_13787 2 235355721 1 2 0.7234 3.178
SNP_25383 4 189927377 1 2 0.3309 3.167
SNP_25290 4 187995996 1 2 0.6327 0.427
SNP_21478 4 40161304 1 2 0.06454 5.066
SNP_12129 2 176643771 1 2 0.9378 1.276
SNP_22809 4 101441465 1 2 0.8111 0.004
3.4 TARGET文件
测试文件,包括:plink的二进制文件和 对应的表型数据文件:
包括2000个个体。
ls TOY_TARGET_DATA.*
TOY_TARGET_DATA.bed TOY_TARGET_DATA.bim TOY_TARGET_DATA.fam TOY_TARGET_DATA.pheno
二进制文件包括:bim,bed和fam文件
表型数据文件:
FID IID Pheno
CAS_1 CAS_1 0.687940475297167
CAS_2 CAS_2 -0.156139175886002
CAS_3 CAS_3 -0.690876014335686
CAS_4 CAS_4 -0.147899250768441
CAS_5 CAS_5 -0.66034095162489
CAS_6 CAS_6 0.0438675950151819
CAS_7 CAS_7 -0.472359275893301
CAS_8 CAS_8 1.49482653529014
CAS_9 CAS_9 1.01876296041241
4. PRSice-2参数介绍
查看帮助文档:
./PRSice_linux
文档大体分为几个部分:
- Base file,基础文件,可以根据指定a1,a2,maf,beta,bp,chr,pvalue等信息
- Target file,目标文件,支持plink二进制文件,可以定义表型,maf质控等
- Dosage,主要是定义运行的资源配置,比如线程、硬盘、内存等(可以省略)
- Clumping,主要是质控Clumping参数,有默认值(可以省略)
- Covariates,定义协变量,包括数字协变量和因子协变量
- P-value Thresholding,P值的阈值定义
- PRSet
- Misc
具体参数介绍:
$ ./PRSice_linux
usage: PRSice [options] <-b base_file> <-t target_file>
Base File:
--a1 Column header containing allele 1 (effective allele)
Default: A1
--a2 Column header containing allele 2 (non-effective allele)
Default: A2
--base | -b Base association file
--base-info Base INFO score filtering. Format should be
<Column name>:<Threshold>. SNPs with info
score less than <Threshold> will be ignored
Column name default: INFO
Threshold default: 0.9
--base-maf Base MAF filtering. Format should be
<Column name>:<Threshold>. SNPs with maf
less than <Threshold> will be ignored. An
additional column can also be added (e.g.
also filter MAF for cases), using the
following format:
<Column name>:<Threshold>,<Column name>:<Threshold>
--beta Whether the test statistic is in the form of
BETA or OR. If set, test statistic is assume
to be in the form of BETA. Mutually exclusive
from --or
--bp Column header containing the SNP coordinate
Default: BP
--chr Column header containing the chromosome
Default: CHR
--index If set, assume the INDEX instead of NAME for
the corresponding columns are provided. Index
should be 0-based (start counting from 0)
--no-default Remove all default options. If set, PRSice
will not set any default column name and you
must manually provide all required columns
(--snp, --stat, --A1, --pvalue)
--or Whether the test statistic is in the form of
BETA or OR. If set, test statistic is assume
to be in the form of OR. Mutually exclusive
from --beta
--pvalue | -p Column header containing the p-value
Default: P
--snp Column header containing the SNP ID
Default: SNP
--stat Column header containing the summary statistic
If --beta is set, default as BETA. Otherwise,
will search for OR or BETA from the header
of the base file
Target File:
--binary-target Indicate whether the target phenotype
is binary or not. Either T or F should be
provided where T represent a binary phenotype.
For multiple phenotypes, the input should be
separated by comma without space.
Default: T if --beta and F if --beta is not
--geno Filter SNPs based on gentype missingness
--info Filter SNPs based on info score. Only used
for imputed target
--keep File containing the sample(s) to be extracted from
the target file. First column should be FID and
the second column should be IID. If --ignore-fid is
set, first column should be IID
Mutually exclusive from --remove
--maf Filter SNPs based on minor allele frequency (MAF)
--nonfounders Keep the nonfounders in the analysis
Note: They will still be excluded from LD calculation
--pheno | -f Phenotype file containing the phenotype(s).
First column must be FID of the samples and
the second column must be IID of the samples.
When --ignore-fid is set, first column must
be the IID of the samples.
Must contain a header if --pheno-col is
specified
--pheno-col | -F Headers of phenotypes to be included from the
phenotype file
--prevalence | -k Prevalence of all binary trait. If provided
will adjust the ascertainment bias of the R2.
Note that when multiple binary trait is found,
prevalence information must be provided for
all of them
--remove File containing the sample(s) to be removed from
the target file. First column should be FID and
the second column should be IID. If --ignore-fid is
set, first column should be IID
Mutually exclusive from --keep
--target | -t Target genotype file. Currently support
both BGEN and binary PLINK format. For
multiple chromosome input, simply substitute
the chromosome number with #. PRSice will
automatically replace # with 1-22
For binary plink format, you can also specify
a seperate fam file by <prefix>,<fam file>
--target-list File containing prefix of target genotype
files. Similar to --target but allow more
flexibility. Do not support external fam file
at the moment
--type File type of the target file. Support bed
(binary plink) and bgen format. Default: bed
Dosage:
--allow-inter Allow the generate of intermediate file. This will
speed up PRSice when using dosage data as clumping
reference and for hard coding PRS calculation
--dose-thres Translate any SNPs with highest genotype probability
less than this threshold to missing call
--hard-thres A hardcall is saved when the distance to the nearest
hardcall is less than the hardcall threshold.
Otherwise a missing code is saved
Default is: 0.1
--hard Use hard coding instead of dosage for PRS construction.
Default is to use dosage instead of hard coding
Clumping:
--clump-kb The distance for clumping in kb
Default: 250
--clump-r2 The R2 threshold for clumping
Default: 0.1 (1mb for PRSet)
--clump-p The p-value threshold use for clumping.
Default: 1
--ld | -L LD reference file. Use for LD calculation. If not
provided, will use the post-filtered target genotype
for LD calculation. Support multiple chromosome input
Please see --target for more information
--ld-dose-thres Translate any SNPs with highest genotype probability
less than this threshold to missing call
--ld-geno Filter SNPs based on genotype missingness
--ld-hard-thres A hardcall is saved when the distance to the nearest
hardcall is less than the hardcall threshold.
Otherwise a missing code is saved
Default is: 0.1
--ld-info Filter SNPs based on info score. Only used
for imputed LD reference
--ld-keep File containing the sample(s) to be extracted from
the LD reference file. First column should be FID and
the second column should be IID. If --ignore-fid is
set, first column should be IID
Mutually exclusive from --ld-remove
No effect if --ld was not provided
--ld-list File containing prefix of LD reference files.
Similar to --ld but allow more
flexibility. Do not support external fam file
at the moment
--ld-maf Filter SNPs based on minor allele frequency
--ld-remove File containing the sample(s) to be removed from
the LD reference file. First column should be FID and
the second column should be IID. If --ignore-fid is
set, first column should be IID
Mutually exclusive from --ld-keep
--ld-type File type of the LD file. Support bed (binary plink)
and bgen format. Default: bed
--no-clump Stop PRSice from performing clumping
--proxy Proxy threshold for index SNP to be considered
as part of the region represented by the clumped
SNP(s). e.g. --proxy 0.8 means the index SNP will
represent region of any clumped SNP(s) that has a
R2>=0.8 even if the index SNP does not physically
locate within the region
Covariate:
--cov | -C Covariate file. First column should be FID and
the second column should be IID. If --ignore-fid
is set, first column should be IID
--cov-col | -c Header of covariates. If not provided, will use
all variables in the covariate file. By adding
@ in front of the string, any numbers within [
and ] will be parsed. E.g. @PC[1-3] will be
read as PC1,PC2,PC3. Discontinuous input are also
supported: @cov[1.3-5] will be parsed as
cov1,cov3,cov4,cov5
--cov-factor Header of categorical covariate(s). Dummy variable
will be automatically generated. Any items in
--cov-factor must also be found in --cov-col
Also accept continuous input (start with @).
P-value Thresholding:
--bar-levels Level of barchart to be plotted. When --fastscore
is set, PRSice will only calculate the PRS for
threshold within the bar level. Levels should be
comma separated without space
--fastscore Only calculate threshold stated in --bar-levels
--no-full By default, PRSice will include the full model,
i.e. p-value threshold = 1. Setting this flag will
disable that behaviour
--interval | -i The step size of the threshold. Default: 5e-05
--lower | -l The starting p-value threshold. Default: 5e-08
--model Genetic model use for regression. The genetic
encoding is based on the base data where the
encoding represent number of the coding allele
Available models include:
add - Additive model, code as 0/1/2 (default)
dom - Dominant model, code as 0/1/1
rec - Recessive model, code as 0/0/1
het - Heterozygous only model, code as 0/1/0
--missing Method to handle missing genotypes. By default,
final scores are averages of valid per-allele
scores with missing genotypes contribute an amount
proportional to imputed allele frequency. To throw
out missing observations instead (decreasing the
denominator in the final average when this happens),
use the 'SET_ZERO' modifier. Alternatively,
you can use the 'CENTER' modifier to shift all scores
to mean zero.
--no-regress Do not perform the regression analysis and simply
output all PRS.
--score Method to calculate the polygenic score.
Available methods include:
avg - Take the average effect size (default)
std - Standardize the effect size
con-std - Standardize the effect size using mean
and sd derived from control samples
sum - Direct summation of the effect size
--upper | -u The final p-value threshold. Default: 0.5
PRSet:
--background String to indicate a background file. This string
should have the format of Name:Type where type can be
bed - 0-based range with 3 column. Chr Start End
range - 1-based range with 3 column. Chr Start End
gene - A file contain a column of gene name
--bed | -B Bed file containing the selected regions.
Name of bed file will be used as the region
identifier. WARNING: Bed file is 0-based
--feature Feature(s) to be included from the gtf file.
Default: exon,CDS,gene,protein_coding.
--full-back Use the whole genome as background for competitive
p-value calculation
--gtf | -g GTF file containing gene boundaries. Required
when --msigdb is used
--msigdb | -m MSIGDB file containing the pathway information.
Require the gtf file
--snp-set Provide a SNP set file containing the snp set(s).
Two different file format is allowed:
SNP list format - A file containing a single
column of SNP ID. Name of the
set will be the file name or
can be provided using
--snp-set File:Name
MSigDB format - Each row represent a single SNP
set with the first column
containing the name of the SNP
set.
--wind-3 Add N base(s) to the 3' region of each feature(s)
--wind-5 Add N base(s) to the 5' region of each feature(s)
Misc:
--all-score Output PRS for ALL threshold. WARNING: This
will generate a huge file
--chr-id Try to construct an RS ID for SNP based on its
chromosome, coordinate, effective allele and
non-effective allele.
e.g. c:L-aBd is translated to:
<chr>:<coordinate>-<effective><noneffective>d
This is always true for target file, whereas for
base file, this is only used if the RS ID
wasn't provided
--exclude File contains SNPs to be excluded from the
analysis
--extract File contains SNPs to be included in the
analysis
--id-delim This parameter causes sample IDs to be parsed as
<FID><delimiter><IID>; the default delimiter
is '_'.
--ignore-fid Ignore FID for all input. When this is set,
first column of all file will be assume to
be IID instead of FID
--keep-ambig Keep ambiguous SNPs. Only use this option
if you are certain that the base and target
has the same A1 and A2 alleles
--logit-perm When performing permutation, still use logistic
regression instead of linear regression. This
will substantially slow down PRSice
--memory Maximum memory usage allowed (in Mb). PRSice will try
its best to honor this setting
--non-cumulate Calculate non-cumulative PRS. PRS will be reset
to 0 for each new P-value threshold instead of
adding up
--out | -o Prefix for all file output
--perm Number of permutation to perform. This swill
generate the empirical p-value. Recommend to
use value larger than 10,000
--print-snp Print all SNPs that remains in the analysis
after clumping is performed. For PRSet, Y
indicate the SNPs falls within the gene set
of interest and N otherwise. If only PRSice
is performed, a single "gene set" called
"Base" will be presented with all entries
marked as Y
--seed | -s Seed used for permutation. If not provided,
system time will be used as seed. When same
seed and same input is provided, same result
can be generated
--thread | -n Number of thread use
--use-ref-maf When specified, missingness imputation will be
performed based on the reference samples
--ultra Ultra aggressive memory usage. When this is enabled
PRSice and PRSet will try to load all genotypes into
memory after clumping is performed. This should
drastically speed up PRSice and PRSet at the expense
of higher memory consumption.
Has no effect for dosage score
--x-range Range of SNPs to be excluded from the whole
analysis. It can either be a single bed file
or a comma seperated list of range. Range must
be in the format of chr:start-end or chr:coordinate
--help | -h Display this help message
Please provide the required parameters
5. 二分类性状计算
5.1 运行代码
代码:
Rscript PRSice.R --dir . --prsice ./PRSice_linux --base TOY_BASE_GWAS.assoc --target TOY_TARGET_DATA --thread 1 --stat OR --binary-target T
- Rscript,是用R语言进行操作
- –dir,默认是当前路径
- –prsice,指定PRSice的执行文件
- –base,是基础数据,这里是GWAS的结果,TOY_BASE_GWAS.assoc
- –target,这里是plink的二进制文件,前缀名
- –thread 1,用1个线程
- –stat OR,这里用的是OR值(二分类性状的OR值,连续性状是Beta值)
- –binary-target T,用的是二分类性状
5.2 运行日志
日志文件:
$ Rscript PRSice.R --dir . --prsice ./PRSice_linux --base TOY_BASE_GWAS.assoc --target TOY_TARGET_DATA --thread 1 --stat OR --binary-target T
PRSice 2.3.3 (2020-08-05)
https://github.com/choishingwan/PRSice
(C) 2016-2020 Shing Wan (Sam) Choi and Paul F. O'Reilly
GNU General Public License v3
If you use PRSice in any published work, please cite:
Choi SW, O'Reilly PF.
PRSice-2: Polygenic Risk Score Software for Biobank-Scale Data.
GigaScience 8, no. 7 (July 1, 2019)
2022-10-21 11:07:18
./PRSice_linux \
--a1 A1 \
--a2 A2 \
--bar-levels 0.001,0.05,0.1,0.2,0.3,0.4,0.5,1 \
--base TOY_BASE_GWAS.assoc \
--binary-target T \
--bp BP \
--chr CHR \
--clump-kb 250kb \
--clump-p 1.000000 \
--clump-r2 0.100000 \
--interval 5e-05 \
--lower 5e-08 \
--num-auto 22 \
--or \
--out PRSice \
--pvalue P \
--seed 2668735456 \
--snp SNP \
--stat OR \
--target TOY_TARGET_DATA \
--thread 1 \
--upper 0.5
Initializing Genotype file: TOY_TARGET_DATA (bed)
Start processing TOY_BASE_GWAS
==================================================
Base file: TOY_BASE_GWAS.assoc
Header of file is:
SNP CHR BP A1 A2 P OR
Reading 100.00%
91062 variant(s) observed in base file, with:
2226 variant(s) located on haploid chromosome
88836 total variant(s) included from base file
Loading Genotype info from target
==================================================
2000 people (1024 male(s), 976 female(s)) observed
2000 founder(s) included
Warning: Currently not support haploid chromosome and sex
chromosomes
88836 variant(s) included
There are a total of 1 phenotype to process
Start performing clumping
Clumping Progress: 100.00%
Number of variant(s) after clumping : 88836
Processing the 1 th phenotype
Phenotype is a binary phenotype
1000 control(s)
1000 case(s)
Start Processing
Processing 100.00%
There are 1 region(s) with p-value less than 1e-5. Please
note that these results are inflated due to the overfitting
inherent in finding the best-fit PRS (but it's still best
to find the best-fit PRS!).
You can use the --perm option (see manual) to calculate an
empirical P-value.
Begin plotting
Current Rscript version = 2.3.3
Plotting Bar Plot
Plotting the high resolution plot
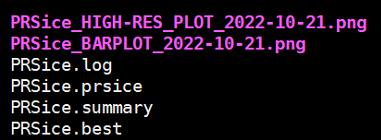
5.3 运行结果
结果文件:
6. 二分类结果解释
6.1 PRSice.price文件
该文件,主要是根据不同Threshold阈值后,选择的SNP个数(Num_SNP)以及对应的解释度(R2)等信息
Pheno Set Threshold R2 P Coefficient Standard.Error Num_SNP
- Base 0.00025005 0.0133696 8.43169e-06 -0.197266 0.0442903 2
- Base 0.00030005 0.00824473 0.000456434 -0.225204 0.0642503 3
- Base 0.00040005 0.0089725 0.000256089 -0.350267 0.0958035 5
- Base 0.00045005 0.0101339 0.000102845 -0.445497 0.114707 6
- Base 0.00065005 0.00532975 0.004775 -0.402003 0.142462 8
- Base 0.00070005 0.00876654 0.00030122 -0.549246 0.151967 9
- Base 0.00080005 0.00233607 0.061455 -0.369219 0.197422 13
- Base 0.00085005 0.00153157 0.129826 -0.342923 0.226384 15
- Base 0.00095005 0.000124324 0.665873 -0.100725 0.233258 16
6.2 PRSice.best文件
这个文件,是每个个体,计算的PRS值
FID IID In_Regression PRS
CAS_1 CAS_1 Yes -0.00599501328
CAS_2 CAS_2 Yes -0.00631017938
CAS_3 CAS_3 Yes -0.00227495325
CAS_4 CAS_4 Yes -0.00204360007
CAS_5 CAS_5 Yes -0.000830676955
CAS_6 CAS_6 Yes -0.00224943517
CAS_7 CAS_7 Yes -0.000687589983
CAS_8 CAS_8 Yes -0.00413102565
CAS_9 CAS_9 Yes 0.00256661049
6.3 PRSice.summy文件
这个文件,是给出最优模型的结果,比如适合的SNP个数,R2,回归系数,P值等信息。
head PRSice.summary
Phenotype Set Threshold PRS.R2 Full.R2 Null.R2 Prevalence Coefficient Standard.Error P Num_SNP
- Base 0.4463 0.0520082 0.0520082 0 - 86.288 9.96331 4.69368e-18 36759
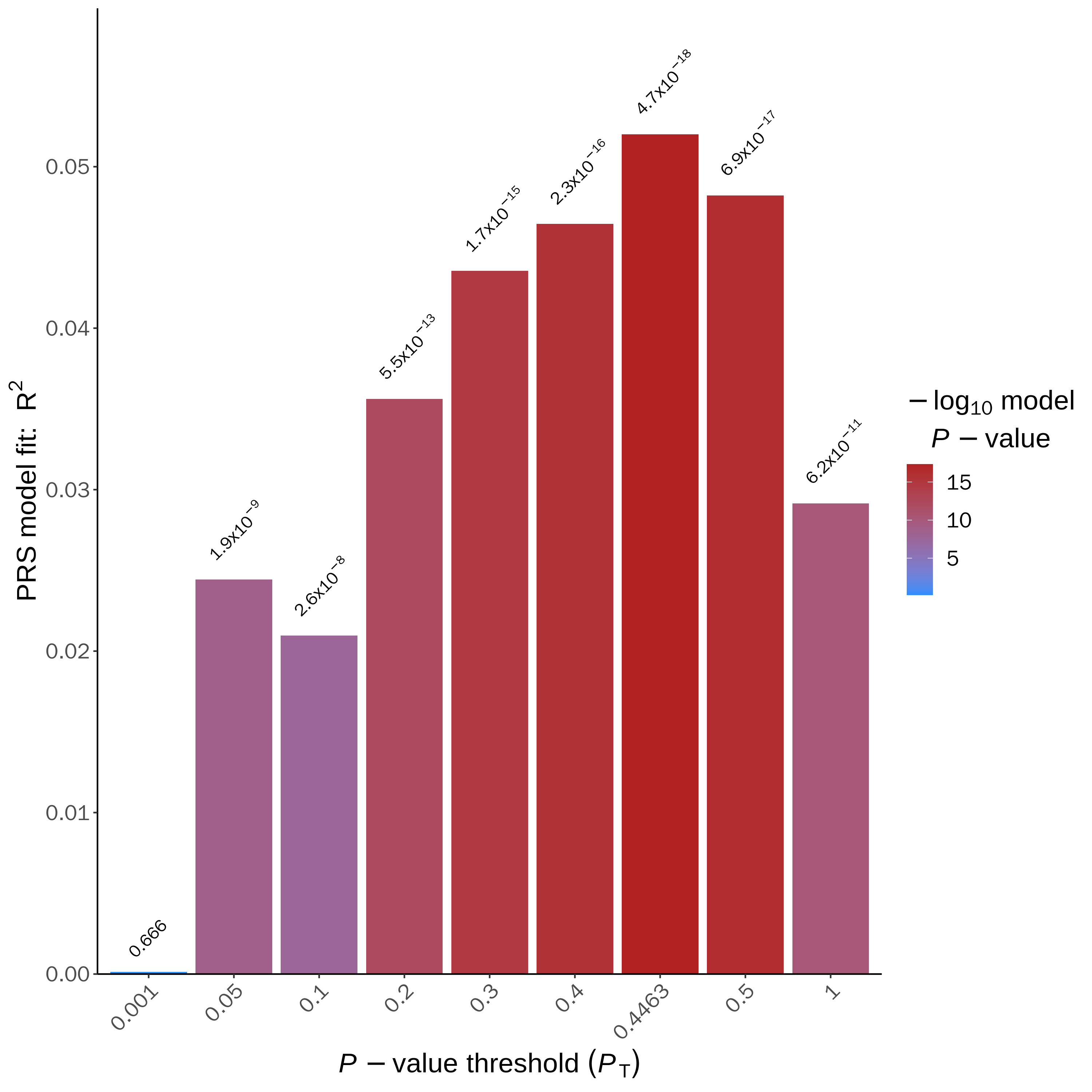
6.4 PRSice_BARPLOT_*.png
这个柱形图,是应用比较广泛的图,X坐标是不同P值,Y坐标是PRS风险得分的解释百分比(R2),柱形图最高的点表示该模型最优,比如下面图中,在P值为0.4463时,模型最优,解释的百分比是5%左右,P值为4.7e-18,极显著。
0.
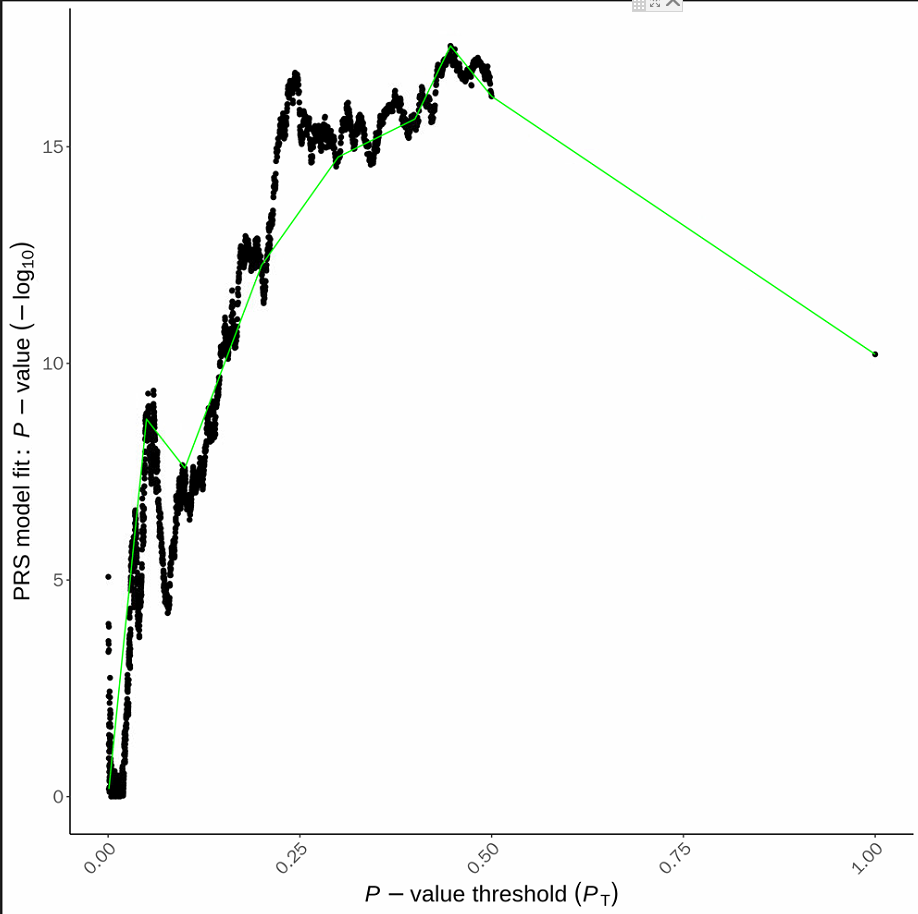
6.5 PRSice_HIGH-RES_PLOT_*.png
下图X坐标是不同的P阈值,Y坐标是显著性(-log转化),可以看到最显著的P的阈值是在0.5左右。
7. 连续性状计算
7.1 运行代码
注意,这里的Base数据是OR,这里强行用连续性状演示一下:
Rscript PRSice.R --dir . --prsice ./PRSice_linux --base TOY_BASE_GWAS.assoc --target TOY_TARGET_DATA --thread 1 --beta --binary-target T
- –beta,这里用的是连续性状
7.2 运行日志
$ Rscript PRSice.R --dir . --prsice ./PRSice_linux --base TOY_BASE_GWAS.assoc --target TOY_TARGET_DATA --thread 1 --beta --binary-target T
PRSice 2.3.3 (2020-08-05)
https://github.com/choishingwan/PRSice
(C) 2016-2020 Shing Wan (Sam) Choi and Paul F. O'Reilly
GNU General Public License v3
If you use PRSice in any published work, please cite:
Choi SW, O'Reilly PF.
PRSice-2: Polygenic Risk Score Software for Biobank-Scale Data.
GigaScience 8, no. 7 (July 1, 2019)
2022-10-21 11:45:37
./PRSice_linux \
--a1 A1 \
--a2 A2 \
--bar-levels 0.001,0.05,0.1,0.2,0.3,0.4,0.5,1 \
--base TOY_BASE_GWAS.assoc \
--beta \
--binary-target T \
--bp BP \
--chr CHR \
--clump-kb 250kb \
--clump-p 1.000000 \
--clump-r2 0.100000 \
--interval 5e-05 \
--lower 5e-08 \
--num-auto 22 \
--out PRSice \
--pvalue P \
--seed 752466145 \
--snp SNP \
--stat OR \
--target TOY_TARGET_DATA \
--thread 1 \
--upper 0.5
Initializing Genotype file: TOY_TARGET_DATA (bed)
Start processing TOY_BASE_GWAS
==================================================
Base file: TOY_BASE_GWAS.assoc
Header of file is:
SNP CHR BP A1 A2 P OR
Reading 100.00%
91062 variant(s) observed in base file, with:
2226 variant(s) located on haploid chromosome
88836 total variant(s) included from base file
Loading Genotype info from target
==================================================
2000 people (1024 male(s), 976 female(s)) observed
2000 founder(s) included
Warning: Currently not support haploid chromosome and sex
chromosomes
88836 variant(s) included
There are a total of 1 phenotype to process
Start performing clumping
Clumping Progress: 100.00%
Number of variant(s) after clumping : 88836
Processing the 1 th phenotype
Phenotype is a binary phenotype
1000 control(s)
1000 case(s)
Start Processing
Processing 100.00%
There are 1 region(s) with p-value between 0.1 and 1e-5
(may not be significant).
Begin plotting
Current Rscript version = 2.3.3
Plotting Bar Plot
Plotting the high resolution plot
7.3 运行结果
8. 连续性状结果
结果形式和二分类性状基本一致。
8.1 PRSice.price文件
该文件,主要是根据不同Threshold阈值后,选择的SNP个数(Num_SNP)以及对应的解释度(R2)等信息
Pheno Set Threshold R2 P Coefficient Standard.Error Num_SNP
- Base 0.00025005 0.00845476 0.000387368 -0.0016371 0.000461343 2
- Base 0.00030005 0.00831682 0.000432057 -0.00243545 0.00069195 3
- Base 0.00040005 0.00882526 0.000289046 -0.00418092 0.00115338 5
- Base 0.00045005 0.00881219 0.000292041 -0.00501347 0.00138407 6
- Base 0.00065005 0.00836554 0.00041563 -0.00650631 0.00184317 8
- Base 0.00070005 0.00836393 0.000416159 -0.00731889 0.00207357 9
- Base 0.00080005 0.00830443 0.000436251 -0.0105346 0.00299523 13
- Base 0.00085005 0.00828742 0.00044217 -0.0121423 0.00345584 15
- Base 0.00095005 0.00763954 0.000739963 -0.0124603 0.00369267 16
6.2 PRSice.best文件
这个文件,是每个个体,计算的PRS值
FID IID In_Regression PRS
CAS_1 CAS_1 Yes 11.5721333
CAS_2 CAS_2 Yes 0.781833333
CAS_3 CAS_3 Yes 14.1177
CAS_4 CAS_4 Yes 0.409866667
CAS_5 CAS_5 Yes 13.9332167
CAS_6 CAS_6 Yes 14.7507667
CAS_7 CAS_7 Yes 1.38896667
CAS_8 CAS_8 Yes 0.447766667
CAS_9 CAS_9 Yes 1.41186667
6.3 PRSice.summy文件
这个文件,是给出最优模型的结果,比如适合的SNP个数,R2,回归系数,P值等信息。
Phenotype Set Threshold PRS.R2 Full.R2 Null.R2 Prevalence Coefficient Standard.Error P Num_SNP
- Base 0.00125005 0.0118796 0.0118796 0 - -0.0280966 0.00668843 2.65998e-05 30
6.4 PRSice_BARPLOT_*.png
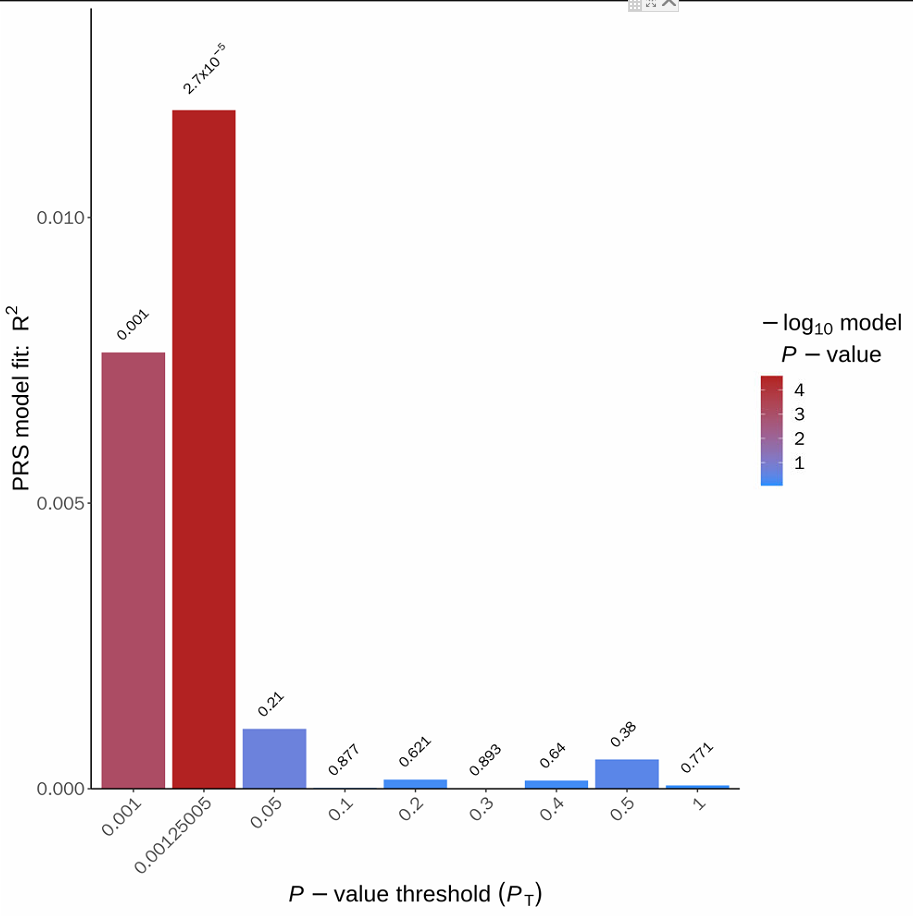
这个柱形图,是应用比较广泛的图,X坐标是不同P值,Y坐标是PRS风险得分的解释百分比(R2),柱形图最高的点表示该模型最优,比如下面图中,在P值为0.00125005时,模型最优,解释的百分比是1.5%左右,P值为2.7e-5,极显著。
6.5 PRSice_HIGH-RES_PLOT_*.png
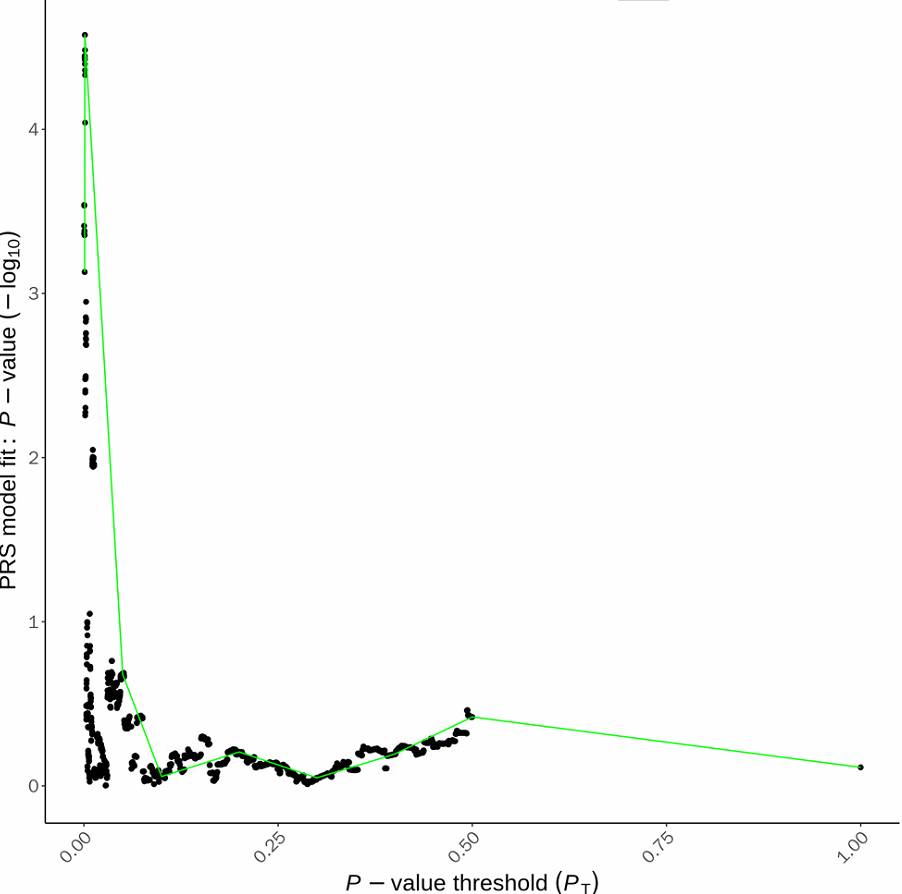
下图X坐标是不同的P阈值,Y坐标是显著性(-log转化),可以看到最显著的P的阈值是在0.5左右。
9. 软件安装好了
下面就是跑程序了。。。

















 6018
6018

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









