







该部分内容仅为从Cellranger到loom文件部分内容,使用的数据集是GSE188711。
Cellranger流程
1.环境部署
进入服务器并设置镜像,如果镜像不行可更换
# 设置镜像
conda config --add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/pkgs/free/
conda config --add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/pkgs/main/
conda config --add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/cloud/bioconda/
conda config --add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/cloud/conda-forge/
conda config --set show_channel_urls yes
创建scRNA环境,用于前期处理
conda create -n scRNA python=3.9
conda activate scRNA
安装sra-tools软件
# conda install -c bioconda sra-tools
# -c是选择默认镜像 可能会存在减慢网速的问题
# 这里的bioconda是一个特定的软件库
# bioconda 可以理解为是一个软件库,-c bioconda 意思是优先从bioconda库来检索适配的软件
# 一般是-c 参数才需要指定特定的库,你没有加 -c参数, 更不需要加bioconda 了
conda install sra-tools
安装Cellranger,官网下载: https://www.10xgenomics.com/support/software/cell-ranger/downloads(请对一下版本)
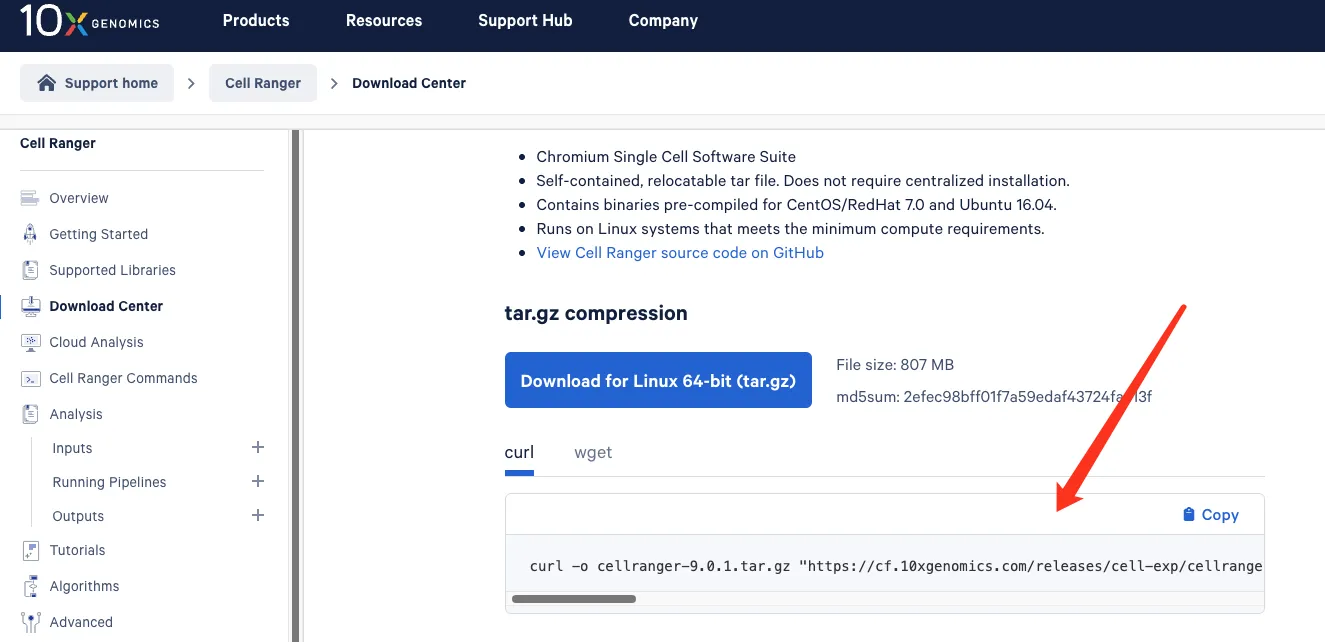
# 在10X官网上下载这个软件
# 下载代码就在网页上有
# 在相应的文件夹中解压缩,软件放置的位置请自行决定
# 笔者这里是创建了biosoft文件夹
# cellranger之前就安装了是9.0.0版本
# 下载好后需要解压
tar -xzvf cellranger-9.0.0.tar.gz
# 在linux或者终端中安装好了之后导入到环境中
# 检查bin中是否存在cellranger
ls -l cellranger-9.0.0
# 临时添加到PATH
export PATH=/home/data/t200558/biosoft/cellranger-9.0.0/bin:$PATH
# which函数确认是否添加成功
which cellranger
下载人类参考基因组其他物种同理,见10X官网
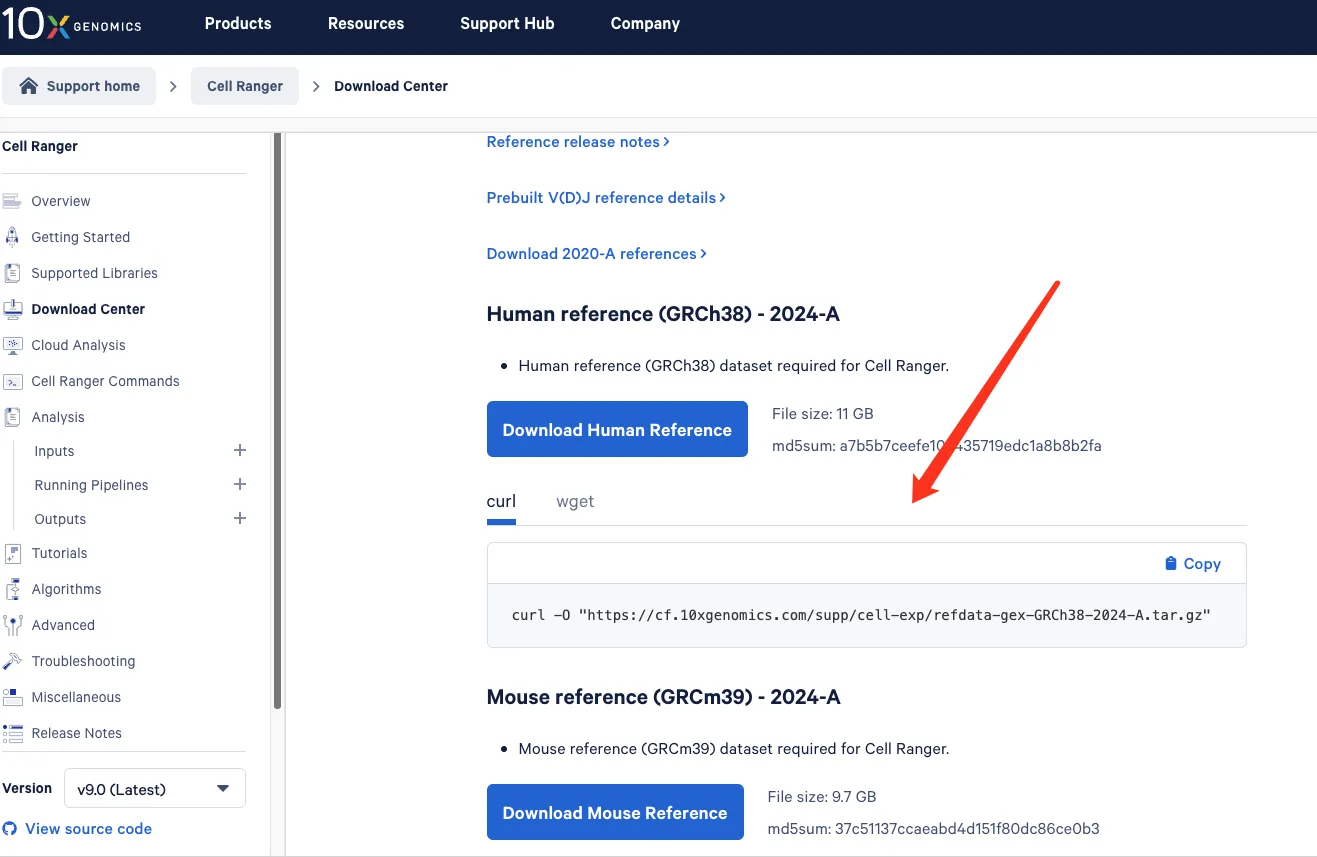
# 同样需要创建一个文件夹放置文件
# 自行设置
nohup curl -O "https://cf.10xgenomics.com/supp/cell-exp/refdata-gex-GRCh38-2024-A.tar.gz" > download.log 2>&1 &
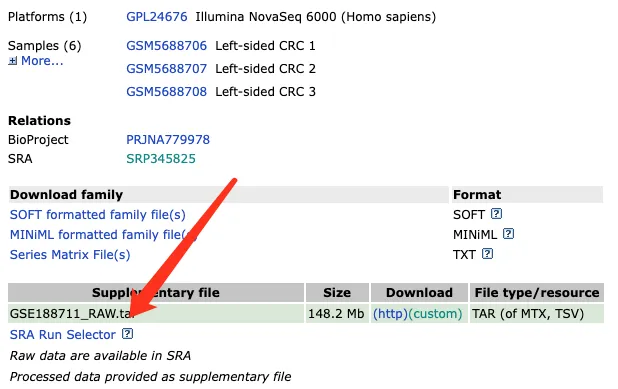
方法一:直接下载,复制SRA Normalized链接即可,但这样的效率肯定是不太行的。
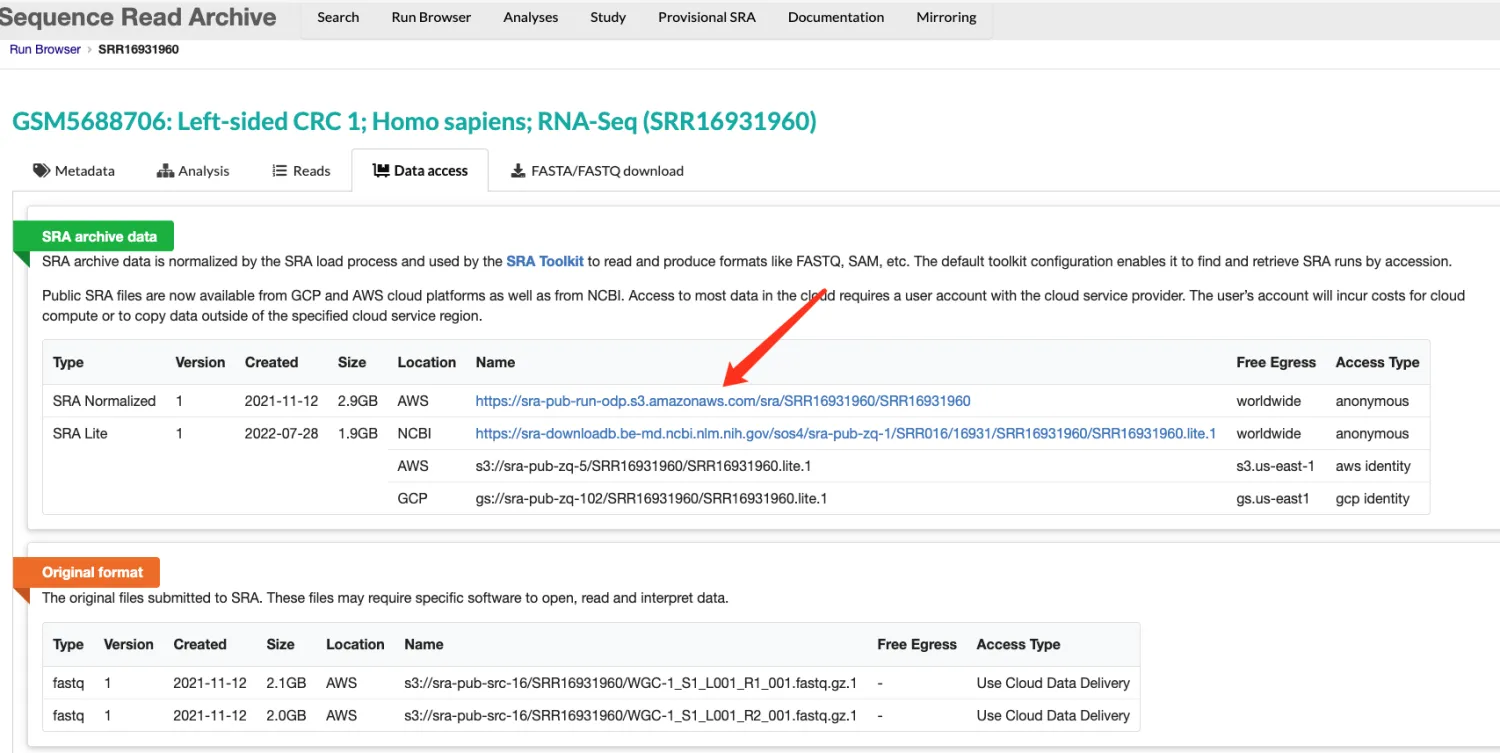
nohup wget https://sra-pub-run-odp.s3.amazonaws.com/sra/SRR16931960/SRR16931960 > download.log > &1 &
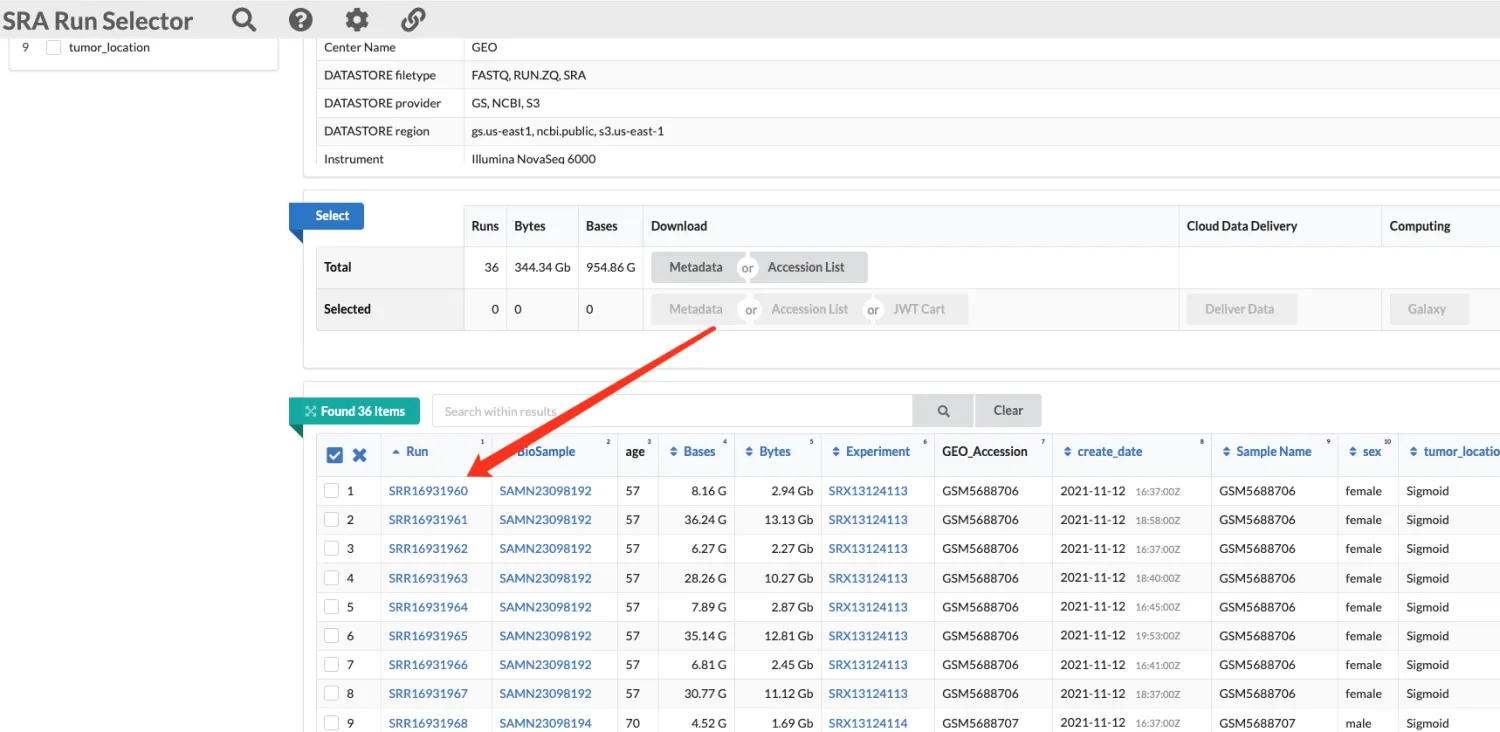
方法二:获得SRR_Acc_List.txt后下载
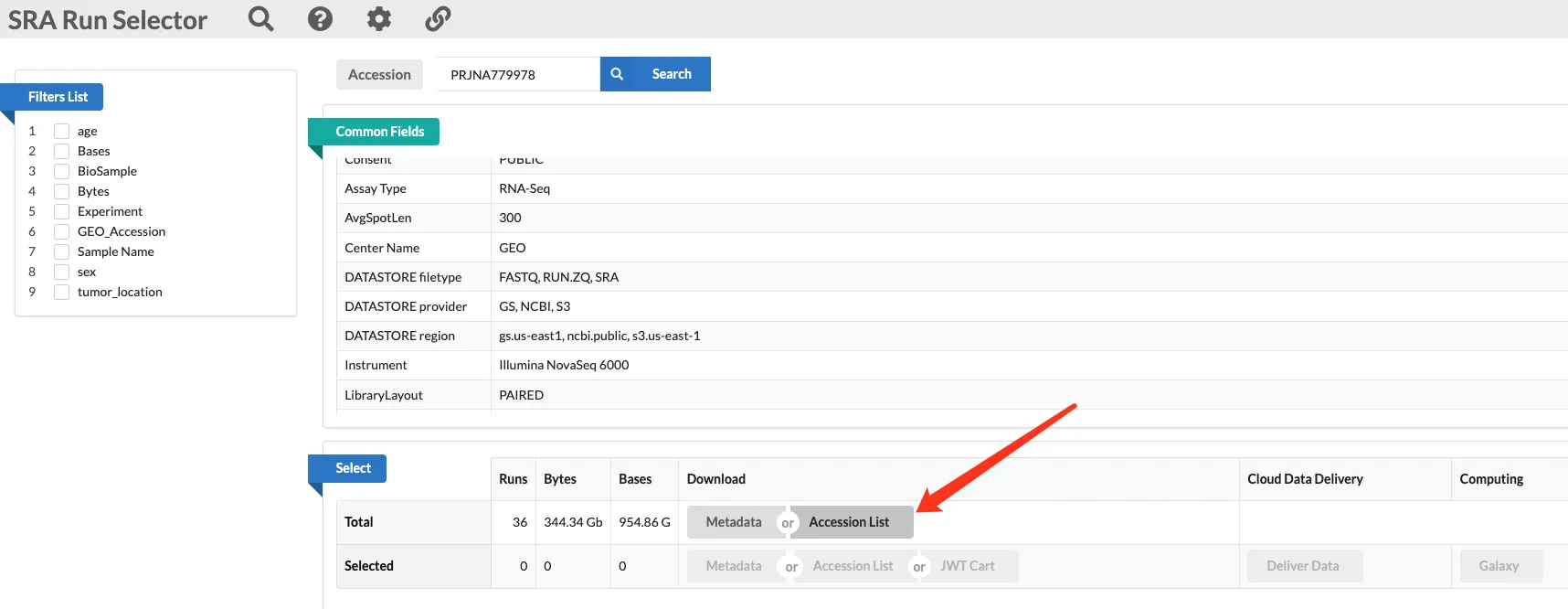
得到一个含有多个SRR号的txt文件并放入指定位置。
使用prefetch下载
prefetch是NCBI SRA工具包提供的命令,用于从 SRA数据库下载数据。笔者这有三种不同的写法,适应不同场景:代码一:使用管道和while read 遍历SRR_Acc_List.txt文件中的每个 SRR ID,依次调用prefetch下载数据,限制最大下载大小为500G。代码二:以相同的方式遍历文件,但是在每次下载完成后打印确认信息。代码三:使用了--option-file选项,通过指定文件来批量下载,适合处理多个文件。这三种方法的区别在于如何指定下载的文件路径,以及是否打印下载状态。通常,prefetch工具适用于从 SRA 服务器下载原始.sra文件,下载速度和稳定性受到 SRA 服务器带宽限制。
# 先需要cd到保存Accession list的文件夹
## 代码一
cat SRR_Acc_List.txt | while read id; do prefetch --max-size 500G "$id"; done
## 代码二
## 遍历 SRR_Acc_List.txt 中的每一行,并下载每个 SRR 文件
cat SRR_Acc_List.txt | while read i
do
prefetch $i --max-size 500G -O `pwd` && echo "** ${i}.sra done **"
done
## 代码三
# 确认当前路径
pwd
# /home/data/t200558/scRNAdata/GSE188711
prefetch \
--option-file /home/data/t200558/scRNAdata/GSE188711/SRR_Acc_List.txt \
-O /home/data/t200558/scRNAdata/GSE188711/ \
--min-size 0 --max-size 500GB
单一样本
nohup prefetch SRR16931965 \
-O /home/data/t200558/scRNAdata/GSE188711/ \
--min-size 0 --max-size 500GB \
> prefetch_SRR16931965.log 2>&1 &
使用kingfisher下载
kingfisher是一个高效的工具,专门设计用于下载大规模的公共数据集(例如 SRA、ENA 等数据库)。它具有以下特点:1. 多线程下载:支持指定下载线程数,能够提高下载速度。2. 数据验证:支持下载后自动校验文件的 MD5 值,确保数据完整性。3. kingfisher更加灵活,适合大规模数据下载任务。需要安装一下aspera,其也是一个高效的下载工具 下载地址:https://www.ibm.com/products/aspera/downloads#cds
在服务器上需要下载linux版本,同时要自行查找aspera的位置。
# 然后cd到根目录下看看是不是存在了.aspera文件夹,有的话表示安装成功
cd && ls -a
# 或者直接查找
sudo find / -name "ascp" 2>/dev/null
# 将aspera软件加入环境变量,并激活
# 这里的位置是按照上面代码查找到的
echo 'export PATH=~/.aspera/connect/bin:$PATH' >> ~/.bashrc
source ~/.bashrc
# 最后检查ascp是不是能用了
ascp --help
pip install kingfisher
conda install pigz
# 此外还需要安装aspera,详细步骤后边
#chck或者直接按照这个进行下载
kingfisher get \
--run-identifiers-list SRR_Acc_List.txt \
-m ena-ascp ena-ftp prefetch \
--download-threads 10 \
--check-md5sums
## nohup后台
nohup bash -c "\
kingfisher get \
--run-identifiers-list SRR16931966 \
-m ena-ascp ena-ftp prefetch \
--download-threads 10 \
--check-md5sums \
1>down_srr_list.log 2>&1" &
## tail -f down_srr_list.log
单一样本
nohup kingfisher get \
-r SRR16931965 \
-m ena-ascp ena-ftp prefetch \
--download-threads 10 \
--check-md5sums \
> down_SRR16931965.log 2>&1 &
这个下载方式要是能够成功的话,其实挺方便的,一行代码解决问题。
使用ascp下载
ascp(Aspera Command Line)是一个高速数据传输工具,用于通过 Aspera 协议加速文件下载,通常用于处理大规模的数据传输。此方法的步骤如下:1. ascp 需要配置私钥文件,这个文件允许通过认证的方式安全地传输文件。2.使用 ascp 命令来下载 SRR 文件,适合需要大文件传输时,ascp 在高速网络条件下提供比传统 FTP 下载更高的速度和稳定性。与 prefetch 相比,ascp 通常在需要加速传输大规模数据时更为有效。
/home/data/t200558/.aspera/connect/bin/ascp -P 33001 -i /home/data/t200558/.aspera/connect/etc/asperaweb_id_dsa.openssh -QT -l 100m -k 1 -d aspera01@download.cncb.ac.cn:gsa-human/HRA007926 /home/data/t200558/scRNAdata/HRA007926
开始下载
# 确定ascp私钥路径
ls ~/.aspera/connect/etc/asperaweb_id_dsa.openssh
# /home/data/t200558/.aspera/connect/etc/asperaweb_id_dsa.openssh
# 提取 ASCP 路径并保存
# 这个路径需要根据不同文件进行修改
awk -F"\t" 'NR > 1 {print $7}' filereport_read_run_PRJNA779978_tsv.txt | grep -o 'ftp.*' | tr ';' '\n' > ascp_paths.txt
# 由于是ftp链接需要修改成aspera能够识别的
# 原来是:ftp.sra.ebi.ac.uk/vol1/fastq/SRR169/062/SRR16931962/SRR16931962_1.fastq.gz
# 修改后:fasp.sra.ebi.ac.uk:/vol1/fastq/SRR169/062/SRR16931962/SRR16931962_1.fastq.gz
sed 's#ftp.sra.ebi.ac.uk#fasp.sra.ebi.ac.uk:#g' ascp_paths.txt > ascp_paths_new.txt
# 使用ascp下载 SRA 文件
nohup bash -c 'cat ascp_paths_new.txt | while read id; do
ascp -QT -l 100m -P 33001 -i /home/data/t200558/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@${id} ./;
done' > download.log 2>&1 &
单一样本(EBI数据库)
EBI的FTP路径通常可以直接转换为Aspera的路径,只需要将服务器地址改为fasp.sra.ebi.ac.uk,并保持后面的路径不变。例如,FTP链接ftp.sra.ebi.ac.uk/vol1/fastq/SRR169/065/SRR16931965/SRR16931965_1.fastq.gz对应的Aspera路径应该是/vol1/fastq/SRR169/065/SRR16931965/SRR16931965_1.fastq.gz,服务器地址为era-fasp@fasp.sra.ebi.ac.uk。
# 下载 SRR16931965_1.fastq.gz
nohup /home/data/t200558/.aspera/connect/bin/ascp \
-P 33001 \
-i /home/data/t200558/.aspera/connect/etc/asperaweb_id_dsa.openssh \
-QT -l 500m -k 1 -d \
--mode=recv \
era-fasp@fasp.sra.ebi.ac.uk:/vol1/fastq/SRR169/065/SRR16931965/SRR16931965_1.fastq.gz \
/home/data/t200558/scRNAdata/GSE188711/SRR16931965 \
> /home/data/t200558/scRNAdata/GSE188711/SRR16931965/ascp_SRR16931965_1.log 2>&1 &
# 下载 SRR16931965_2.fastq.gz
nohup /home/data/t200558/.aspera/connect/bin/ascp \
-P 33001 \
-i /home/data/t200558/.aspera/connect/etc/asperaweb_id_dsa.openssh \
-QT -l 500m -k 1 -d \
--mode=recv \
era-fasp@fasp.sra.ebi.ac.uk:/vol1/fastq/SRR169/065/SRR16931965/SRR16931965_2.fastq.gz \
/home/data/t200558/scRNAdata/GSE188711/SRR16931965 \
> /home/data/t200558/scRNAdata/GSE188711/SRR16931965/ascp_SRR16931965_2.log 2>&1 &
3.md5sum校验和未压缩文件压缩
有时候需要对下载的文件进行校验和压缩
md5sum SRR16931961_1.fastq.gz
md5sum SRR16931961_1.fastq # 若已删除可跳过
# 方法一 gzip
# 压缩单个文件(生成 SRR16931961_1.fastq.gz,并删除原文件)
gzip SRR16931961_1.fastq
# 批量压缩所有未压缩的 .fastq 文件
gzip SRR16931961_*.fastq
# 方法二 pigz
# 需要先安装pigz
# 并行压缩文件(例如使用 8 个线程)
pigz -k -p 8 SRR16931961_1.fastq SRR16931961_2.fastq
4.文件重命名
由于下载的数据已经是fastq文件了,因此可以直接进行count细胞定量。如果下载的内容并不是fastq文件则需要通过mkfastq或者bcl2fastq进行处理。 重命名格式:SampleName_S1_L001_[R/I][1/2]_001.fastq.gz (I1文件可以没有)
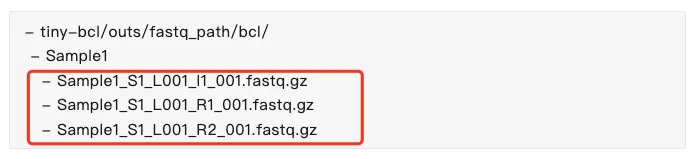
# 创建文件夹
mkdir -p linked_fastq
# 处理所有 *_1.fastq.gz → R1
ls *_1.fastq.gz | while read file; do
sample=$(basename "$file" | cut -d'_' -f1)
ln -sf "$(realpath "$file")" "linked_fastq/${sample}_S1_L001_R1_001.fastq.gz"
done
# 处理所有 *_2.fastq.gz → R2
ls *_2.fastq.gz | while read file; do
sample=$(basename "$file" | cut -d'_' -f1)
ln -sf "$(realpath "$file")" "linked_fastq/${sample}_S1_L001_R2_001.fastq.gz"
done
# 清除其他冗余文件之后之间修改文件名称
# 重命名 *_1.fastq.gz → R1
for file in *_1.fastq.gz; do
sample=$(basename "$file" | cut -d'_' -f1)
mv "$file" "${sample}_S1_L001_R1_001.fastq.gz"
done
# 重命名 *_2.fastq.gz → R2
for file in *_2.fastq.gz; do
sample=$(basename "$file" | cut -d'_' -f1)
mv "$file" "${sample}_S1_L001_R2_001.fastq.gz"
done
5.先按照sample进行归类放置
对照官网信息进行文件夹的创建和移动
# 创建sample名称的文件夹
mkdir -p GSM56887{06..11}
# 移动样本进入相应文件夹
mv SRR169319{68..75}_*.fastq.gz GSM5688707/
mv SRR169319{76..79}_*.fastq.gz GSM5688708/
mv SRR169319{80..87}_*.fastq.gz GSM5688709/
mv SRR169319{88..91}_*.fastq.gz GSM5688710/
mv SRR169319{92..95}_*.fastq.gz GSM5688711/
# 其他文件自行改名
6.再次按照样本进行修改
# 获取当前文件夹名称(例如 GSM5688706)
folder_name=$(basename "$PWD")
# 提取所有唯一的 HRR 前缀,并生成 S1/S2/S3... 的映射
declare -A hr_mapping
counter=1
for file in SRR*.fastq.gz; do
# 提取 SRR 前缀(例如 GSM5688706)
hr_prefix=$(echo "$file" | grep -oE '^SRR[0-9]+' | head -1)
# 去重并分配 S 编号
if [ -n "$hr_prefix" ] && [ -z "${hr_mapping[$hr_prefix]}" ]; then
hr_mapping["$hr_prefix"]="S$counter"
((counter++))
fi
done
# 重命名文件
for file in SRR*.fastq.gz; do
# 分解文件名各部分
hr_prefix=$(echo "$file" | grep -oE '^SRR[0-9]+' | head -1)
s_id="${hr_mapping[$hr_prefix]}" # 获取新 S 编号(如 S1)
# 提取剩余部分,并移除原 S1 字段(第一个下划线后的内容)
rest_part=$(echo "$file" | sed "s/^${hr_prefix}_//")
rest_part_modified=$(echo "$rest_part" | cut -d'_' -f2-) # 去掉原 S1
# 构建新文件名(格式:SRR16931960_S1_L001_R1_001.fastq.gz)
new_name="${folder_name}_${s_id}_${rest_part_modified}"
# 执行重命名(安全模式,避免覆盖)
mv -nv "$file" "$new_name"
done
7.移动文件最分析前最后准备
# 创建新文件夹
mkdir merged_GSM
# 移动文件到该文件夹中
find GSM* -type f -name "*.fastq.gz" -exec mv -v {} merged_GSM/ \;
# 查看所有以fastq.gz结尾的文件,并统计数量
find merged_GSM -type f -name "*.fastq.gz" | wc -l
# 删除GSM开头的文件夹
rm -rv GSM56887{06..11}
8.Cellranger流程
建议一个个样本分析,分析过程很占空间
# 查看特定文件夹所占内存
du -sh ../../scRNAdata/
# 获得所有sample名称,进入merged_HRS文件夹提取
ls *.fastq.gz | cut -d'_' -f1 | sort -u > ../samples.txt
# 临时添加到PATH
export PATH=/home/data/t200558/biosoft/cellranger-9.0.0/bin:$PATH
# which函数确认是否添加成功
which cellranger
# 设定参考基因组
ref="/home/data/t200558/reference/GRCh38/refdata-gex-GRCh38-2024-A"
# 设定fastq文件路径
fastqs="/home/data/t200558/scRNAdata/GSE188711/merged_GSM"
# 输出文件存放路径
#output="/home/data/t200558/scRNAdata/GSE188711"
# 样本名字列表
samples=("GSM5688711") # 这里列出所有样本名字
for sample in "${samples[@]}"
do
echo "正在处理样本:$sample"
nohup cellranger count \
--id="$sample" \
--transcriptome="$ref" \
--fastqs="$fastqs" \
--sample="$sample" \
--create-bam=true \
--nosecondary \
--localcores=4 \
--localmem=30 \
> "${sample}.log" 2>&1 &
done
velocyto分析流程
1.创建velocyto环境
# 创建环境
conda create -n velocyto
conda activate velocyto
# 安装velocyto及其依赖库
conda install numpy scipy cython numba matplotlib scikit-learn h5py click
pip install velocyto
2.运行velocyto分析前准备
需要下载基因组注释文件(.gtf),可以从GENCODE何Ensembl中下载,也可以从Cellranger官网下载 此外,需要下载重复序列元件的注释信息,velocyto提供了链接可以直接进去UCSC下载界面
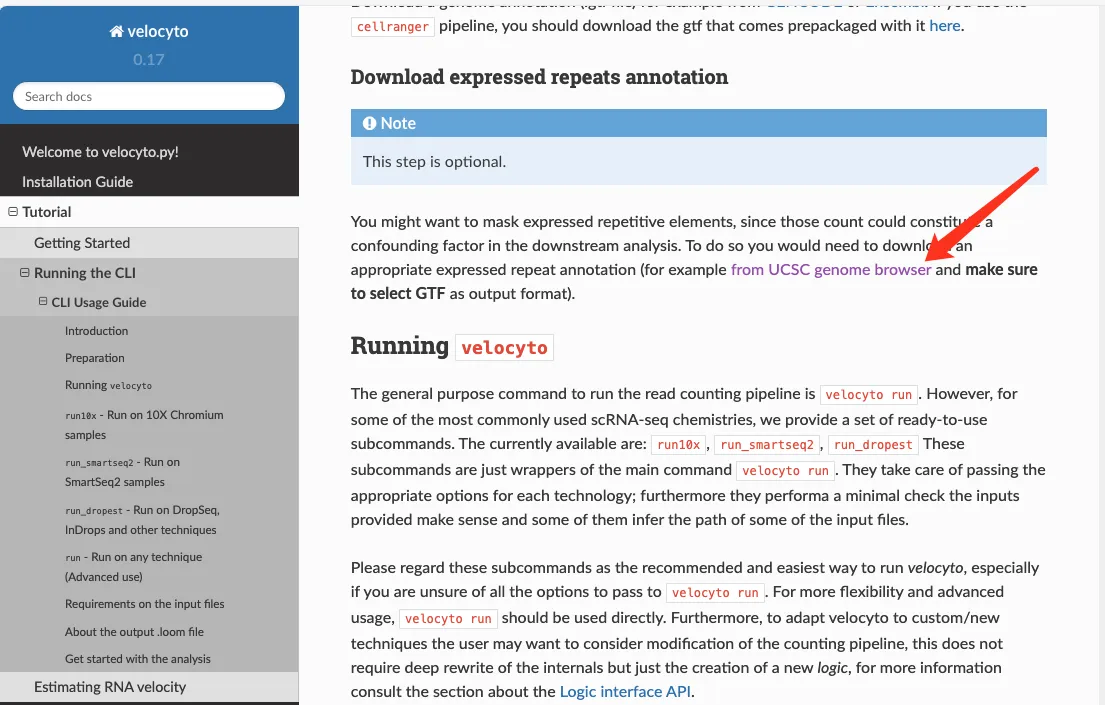
设定参数之后点击Get output获取文件。
但笔者试过之后发现下载的数据有点问题,暂时没法解决。 因此找了简书UP主的内容:https://www.jianshu.com/p/70c19748ac1a,里面有百度云链接。
将bam或sam文件整合生成loom文件
velocyto run10x -m repeat_msk.gtf mypath/sample01 somepath/refdata-cellranger-mm10-1.2.0/genes/genes.gtf
-s, --metadatatable FILE 样本的注释文件,csv格式,行是样本,列是注释信息。(跟seurat,monocle等R包中的metadata格式是一样的)
-m, --mask FILE 去掉一些重复表达的基因, .gtf文件,可以从UCSE gene browser上下载
-l, --logic TEXT 用于筛选的逻辑值(这个参数还没有研究过)
-M, --multimap 考虑非唯一映射(作者不建议这样)
-@, --samtools-threads INTEGER 用samtools对bam文件进行sort时用到的进程数
--samtools-memory INTEGER 每个进程分配的内存(单位Mb)
-t, --dtype TEXT loom文件层的dtype。如果每个细胞的每个基因超过6000个molecules/reads,这个参数建议设置为32。(默认是16)
genes.gtf是cellranger管道提供的基因组注释文件。repeat_msk.gtf是在准备部分中描述的重复掩码文件。mypath/sample01是cellranger输出文件包含了outs、outs/analys和outs/filtered_gene_bc_matrices的文件夹。
# 设定参考基因组
ref="/home/data/t200558/reference/genes.gtf"
# 设定注释文件路径
anno="/home/data/t200558/reference/UCSC_repeats_Anno/human_allTracks_new.gtf"
# 输入文件存放路径,文件夹名称要及时修改
input="/home/data/t200558/scRNAdata/GSE188711/GSM5688706/"
# 正式运行
nohup velocyto run10x -m $anno $input $ref > run_GSM5688706.log 2>&1 &
# velocyto run10x -m $anno $input $ref
将多个loom文件合并到一个文件中,需要在linux/终端中运行
conda activate velocyto
pip install ipykernel
# 其他的内核注册就按照其他的名称来
python -m ipykernel install --user --name velocyto --display-name "Python(velocyto)"
conda install anndata scipy loompy
pip install numpy==1.23 # numpy是1.23版本
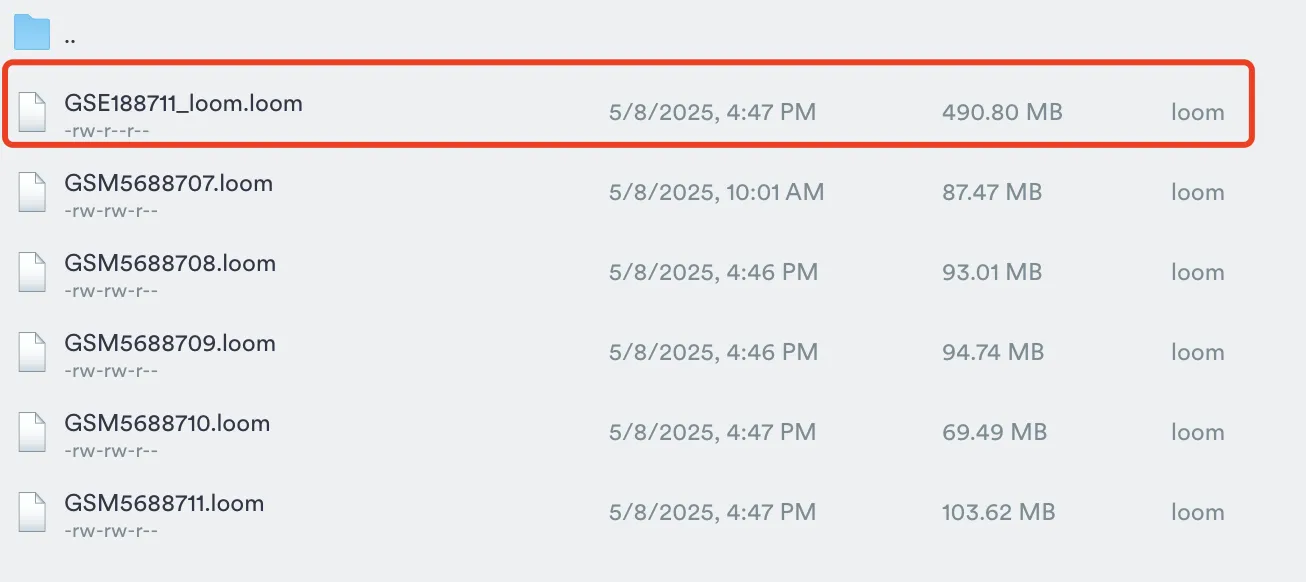
使用loompy进行合并数据,同时笔者这里GSM568806的loom文件没有完成,因此仅合并剩下的5个loom文件。
# 标准格式loompy.combine(files, output_filename, key="Accession")
import loompy
files = [
"GSM5688707.loom",
"GSM5688708.loom",
"GSM5688709.loom",
"GSM5688710.loom",
"GSM5688711.loom"
]
loompy.combine(files, "GSE188711_loom.loom", key="Accession")
会得到一个整合好后的loom文件
3.SeuratV5数据转换为h5ad数据
选择已经降维聚类分群的数据进行h5ad转化,也可以不转化直接使用velocyto.R
R语言环境
rm(list = ls())
library(Seurat)
library(qs)
library(reticulate)
library(hdf5r)
# devtools::install_github("cellgeni/sceasy")
library(sceasy)
library(BiocParallel)
register(MulticoreParam(workers = 4, progressbar = TRUE))
scRNA_V5 <- qread("CD4+t_final.qs")
scRNA_V5
配置python环境(linux/终端)
conda create -n sceasy python=3.9
conda activate sceasy
conda install anndata scipy loompy
R语言转换
# 在R语言中加载python环境
use_condaenv('sceasy')
loompy <- reticulate::import('loompy')
# Seurat to AnnData
scRNA_V5[["RNA"]] <- as(scRNA_V5[["RNA"]], "Assay")
sceasy::convertFormat(scRNA_V5, from="seurat", to="anndata",
outFile='CD4+t_final.h5ad')
在jupyter lab中打开h5ad文件确认一下,需要先注册内核
# 加载库
import scanpy as sc
import os
# 确认路径
os.getcwd()
# 读取数据
adata = sc.read_h5ad('CD4+t_final.h5ad')
adata
参考资料:
-
10xgenomics-cellranger:https://www.10xgenomics.com/support/software/cell-ranger/latest/tutorials/cr-tutorial-in
-
velocyto:https://github.com/velocyto-team/velocyto.R https://github.com/velocyto-team/velocyto.py
-
生信技能树:https://mp.weixin.qq.com/s/X1SYGr2zv-b7hSppNC_22Q https://mp.weixin.qq.com/s/X1SYGr2zv-b7hSppNC_22Q https://mp.weixin.qq.com/s/CQG0QUXrS3hf3OTL0inuZQ https://mp.weixin.qq.com/s/TJsdj7qesZGlvmEkNLAy0A
-
生信菜鸟团:https://mp.weixin.qq.com/s/lvooimNKTQ8U9_2t2SuweQ https://mp.weixin.qq.com/s/AVqv07swFvjl6OCnLwwLPA
-
老俊俊的生信笔记:https://mp.weixin.qq.com/s/1bmR6FC_PD3pzo8d3TrYdA https://mp.weixin.qq.com/s/q_aTuN3S82d6B2qsVz7TVw
-
scRNA velocity: https://github.com/hamidghaedi/scRNA_velocity_analysis
注:若对内容有疑惑或者有发现明确错误的朋友,请联系后台(欢迎交流)。更多相关内容可关注公众号:生信方舟 。

















 3240
3240

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









