







 本文介绍了如何使用R包Goplot进行富集分析可视化,包括从数据准备、差异分析到使用clusterProfiler和enrichGO进行基因富集分析,以及各种图表类型的创建过程。
本文介绍了如何使用R包Goplot进行富集分析可视化,包括从数据准备、差异分析到使用clusterProfiler和enrichGO进行基因富集分析,以及各种图表类型的创建过程。
本文目录:
之前的推文已经给大家详细介绍了clusterprofiler的使用方法以及enrichplot的用法:
今天继续给大家介绍Goplot包进行富集分析可视化,这个R包的图形有一些独到之处,经常在文献中出现,有人说这个R包数据准备很难,如果你能耐心跟着教程走一遍,我觉得这个事情会变得简单。
准备数据
用gse87466这个GEO的数据做演示,下载整理的过程这次就不演示了。数据可在粉丝QQ群免费下载。
load(file = "G:/easyTCGA_test/gse87466.Rdata")
这是一个炎症性肠病的数据集,一共108个样本,21个normal,87个uc(ulcerative colitis)。
exprSet[1:4,1:4]
## GSM2332098 GSM2332099 GSM2332100
## IGK@ /// IGKC 13.86197 13.76880 13.95740
## 13.95740 13.92619 13.79664
## IGL@ 13.73797 13.61266 13.86197
## IGH@ /// IGHA1 /// IGHA2 /// LOC100126583 13.79664 13.16844 13.76880
## GSM2332101
## IGK@ /// IGKC 13.95740
## 13.86197
## IGL@ 13.76880
## IGH@ /// IGHA1 /// IGHA2 /// LOC100126583 13.73797
group <- factor(group_list,levels = c("normal","UC"))
table(group)
## group
## normal UC
## 21 87
首先对这个数据做下差异分析,也是用easyTCGA包,1行代码即可,基因芯片数据也是支持的,并且它会自动检测需不需要进行log2转换,如果是count矩阵,会自动使用DESeq2、limma、edgeR进行差异分析,如果不是,会自动进行wilcoxon和limma的差异分析:
library(easyTCGA)
diff_res <- diff_analysis(exprset = exprSet
, group = group
, is_count = F # 不是count数据
, logFC_cut = 0 # 可以直接筛选结果
, pvalue_cut = 1
)
## log2 transform not needed
## => Running limma
## => Running wilcoxon test
## => Analysis done.
# limma的结果
diff_limma <- diff_res$deg_limma
# 多个gene symbol的直接删除,方便演示
diff_limma <- diff_limma[!grepl("/",diff_limma$genesymbol),]
head(diff_limma)
## logFC AveExpr t P.Value adj.P.Val B
## SLC6A14 5.024103 9.413107 21.56440 4.104849e-41 8.514279e-37 82.58182
## LOC389023 -3.550396 5.541681 -21.01057 4.054400e-40 4.204818e-36 80.36199
## SLC23A1 -2.473180 5.649224 -17.88487 3.378001e-34 2.335550e-30 67.08748
## DUOX2 4.911030 9.916299 17.37129 3.569259e-33 1.850839e-29 64.78265
## DPP10 -1.910958 3.991413 -16.98863 2.113068e-32 7.304876e-29 63.04259
## TIMP1 2.125930 11.402645 16.88534 3.425860e-32 1.015131e-28 62.56956
## genesymbol
## SLC6A14 SLC6A14
## LOC389023 LOC389023
## SLC23A1 SLC23A1
## DUOX2 DUOX2
## DPP10 DPP10
## TIMP1 TIMP1
选取logFC > 1 & adj.P.Val<0.01 的基因作为差异基因进行后续的ORA分析:
deg_limma <- diff_limma[abs(diff_limma$logFC)>1 & diff_limma$adj.P.Val<0.01,]
deg_genes <- deg_limma$genesymbol
length(deg_genes)
## [1] 1192
head(deg_genes)
## [1] "SLC6A14" "LOC389023" "SLC23A1" "DUOX2" "DPP10" "TIMP1"
1192个差异基因等下用于ORA富集分析。
富集分析
富集分析首选clusterProfiler,没有之一!简单,好用!
富集分析最好用ENTREZID进行,但其实不转换也可以进行,富集分析时会给你转换,你只要指定类型即可,这里是因为enrichGO富集分析会借助Org注释包进行,里面含有多种不同的基因ID,它可以自动帮你进行转换,如果没有使用Org注释包的富集分析函数就只能用ENTREZID。
首先进行ORA:
suppressMessages(library(clusterProfiler))
ora_res <- enrichGO(gene = deg_genes,
OrgDb = "org.Hs.eg.db",
keyType = "SYMBOL",#这里指定ID类型
ont = "ALL", # "BP", "MF", "CC"
pvalueCutoff = 0.01,
pAdjustMethod = "BH",
qvalueCutoff = 0.01,
minGSSize = 30,# 最少的基因数量
maxGSSize = 300, # 最大的基因数量
readable = T # 把ENTREZID转换为SYMBOL
)
##
head(ora_res,6)
## ONTOLOGY ID Description GeneRatio BgRatio
## GO:0097530 BP GO:0097530 granulocyte migration 52/1002 158/18903
## GO:0030595 BP GO:0030595 leukocyte chemotaxis 62/1002 240/18903
## GO:0071621 BP GO:0071621 granulocyte chemotaxis 46/1002 130/18903
## GO:1990266 BP GO:1990266 neutrophil migration 46/1002 130/18903
## GO:0030593 BP GO:0030593 neutrophil chemotaxis 41/1002 106/18903
## GO:0097529 BP GO:0097529 myeloid leukocyte migration 59/1002 242/18903
## pvalue p.adjust qvalue
## GO:0097530 1.187840e-27 3.540950e-24 2.413190e-24
## GO:0030595 3.363321e-26 2.825568e-23 1.925650e-23
## GO:0071621 3.791436e-26 2.825568e-23 1.925650e-23
## GO:1990266 3.791436e-26 2.825568e-23 1.925650e-23
## GO:0030593 3.418361e-25 2.038027e-22 1.388934e-22
## GO:0097529 1.395870e-23 6.935145e-21 4.726365e-21
转换数据
使用Goplot进行可视化需要特定格式的数据,需要提前准备好富集分析结果和差异分析结果,我们已经有了富集分析结果ora_res和差异分析结果deg_limma,转换起来就很简单了。
但是这个函数封装的很过度,只要term一多,就会很难看,而且没有给出限制的函数,所以我们提前筛选一下ora_res:
# 在BP, MF , CC各选20个用于演示
res_go <- ora_res %>%
group_by(ONTOLOGY) %>%
slice(1:20)
dim(res_go)
## [1] 58 10
然后把富集分析结果中的几个列名改成它需要的列名,还有差异分析结果的列名也改一下,把这两个结果放到一个list中:
res_go <- res_go@result
names(res_go)[c(1,3,7,9)] <- c("Category","Term","adj_pval","Genes")
res_go$Genes <- gsub("\\/",", ",res_go$Genes)
names(deg_limma)[7] <- "ID"
go_list <- list(resGo = res_go,
degLimma = deg_limma
)
然后就可以使用circle_dat进行转换了:
library(GOplot)
## Loading required package: ggplot2
## Loading required package: ggdendro
## Loading required package: gridExtra
## Loading required package: RColorBrewer
# 转换数据,其实就是一个数据框
circ <- circle_dat(go_list$resGo, go_list$degLimma)
dim(circ)
## [1] 1931 8
head(circ)
## category ID term count genes logFC adj_pval
## 1 BP GO:0097530 granulocyte migration 52 CXCL1 3.761505 3.54095e-24
## 2 BP GO:0097530 granulocyte migration 52 CXCL3 2.472198 3.54095e-24
## 3 BP GO:0097530 granulocyte migration 52 S100A8 4.256463 3.54095e-24
## 4 BP GO:0097530 granulocyte migration 52 ANXA1 1.815406 3.54095e-24
## 5 BP GO:0097530 granulocyte migration 52 CXCL2 2.977254 3.54095e-24
## 6 BP GO:0097530 granulocyte migration 52 CXCL9 2.915224 3.54095e-24
## zscore
## 1 5.269652
## 2 5.269652
## 3 5.269652
## 4 5.269652
## 5 5.269652
## 6 5.269652
后面就可以画图了,我已经看过源码了,竟然都是ggplot2画出来的,不过是进行了封装,简化了自己整理数据并画图的过程,真的是很6!
条形图
GOBar(subset(circ, category == 'BP'))
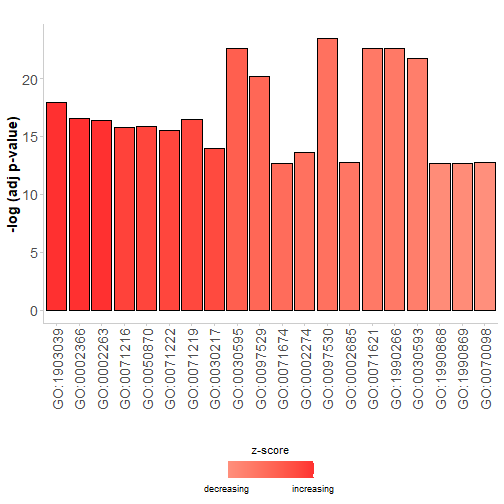
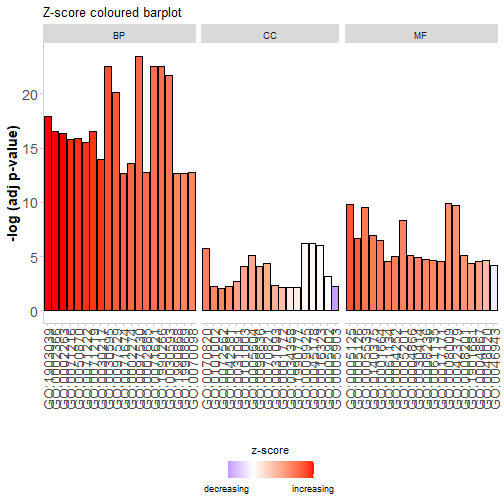
分面条形图,可以更改标题、颜色等:
GOBar(circ, display = 'multiple',
title = 'Z-score coloured barplot',
zsc.col = c('red', 'white', 'blue'))
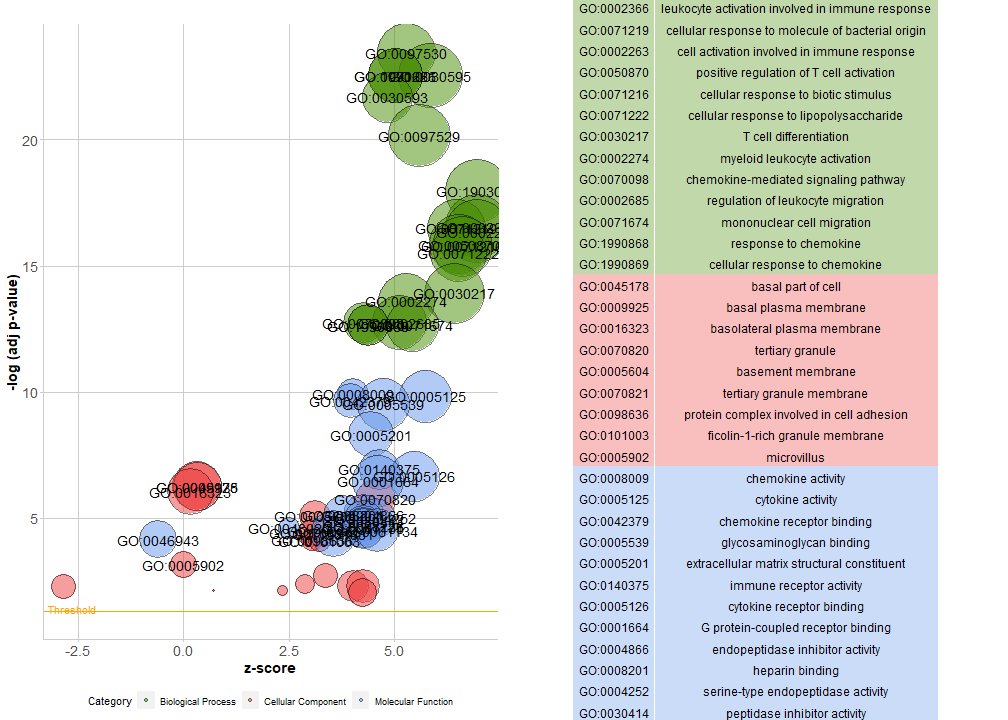
气泡图
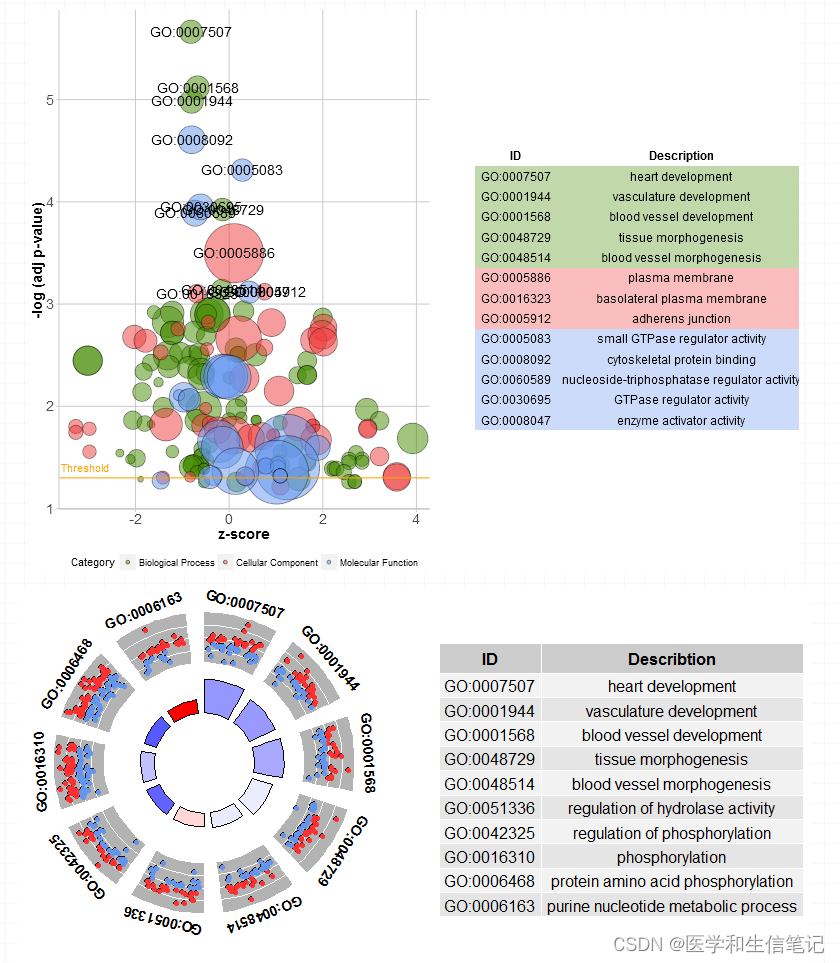
把BP,CC,MF都画在一张图里,并添加条目信息表格:
GOBubble(circ, labels = 3)
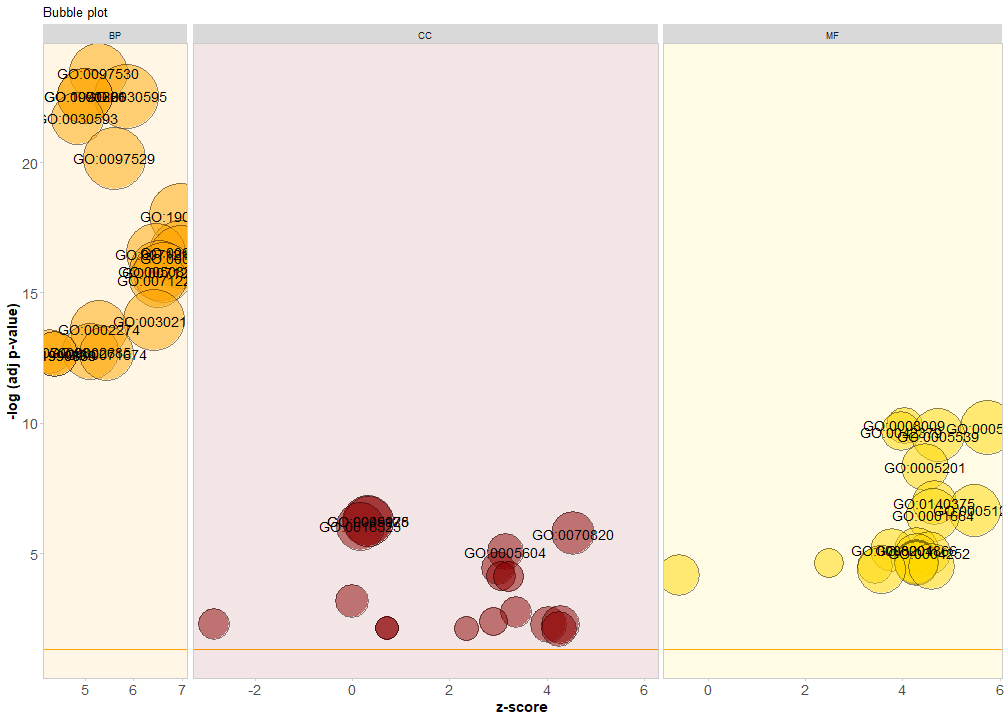
当然也可以分面画,可以更改标题、颜色等:
GOBubble(circ,
title = 'Bubble plot',
colour = c('orange', 'darkred', 'gold'),
display = 'multiple',
bg.col = T, #是否显示背景色
labels = 5) #显示标签的阈值
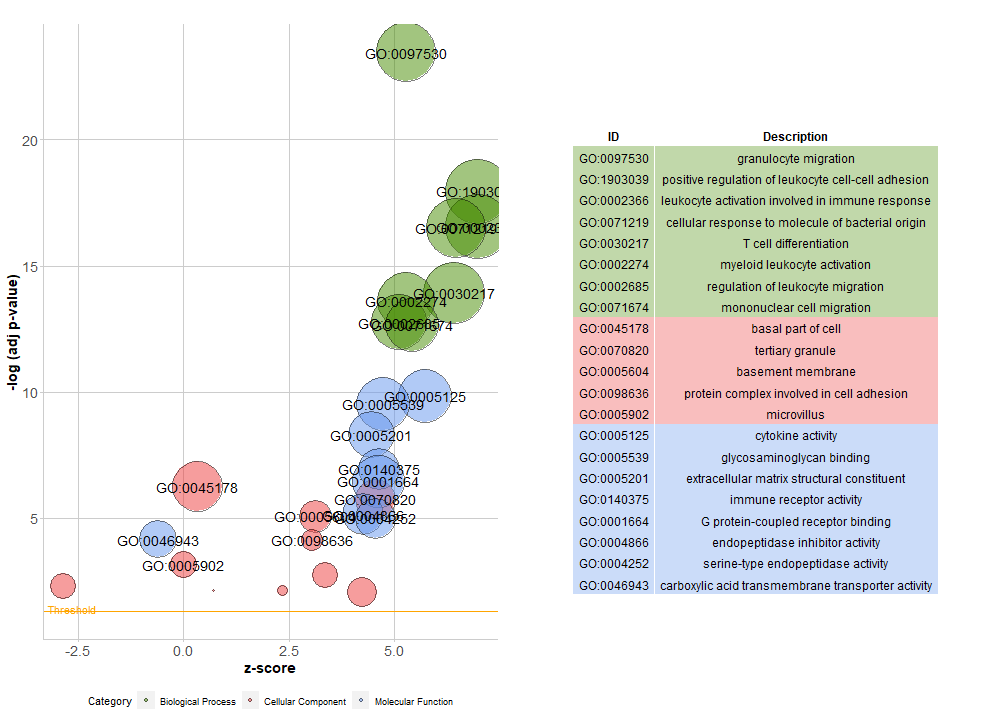
也提供了一个可以去冗余的函数:
# Reduce redundant terms with a gene overlap >= 0.75...
reduced_circ <- reduce_overlap(circ, overlap = 0.75)
GOBubble(reduced_circ, labels = 2.8)
八卦图
作者说这个图就是为了着重显示z-score,怎么计算可以参考:https://wencke.github.io/。反正不用自己算,转换数据时已经帮我们算好了。
GOCircle(circ,
title="GO circle plot",
nsub=10, #展示哪些条目,也可以使用条目名字组成的字符向量
rad1=2, #里面圆环的半径
rad2=3, #外面圆环的半径
table.legend = T, #是否展示表格
zsc.col=c("red","white","blue"), # zscore的颜色
lfc.col=c("steelblue","orange"), # logfc颜色
label.size=5, #条目标签大小
label.fontface="bold" #条目标签样式
)
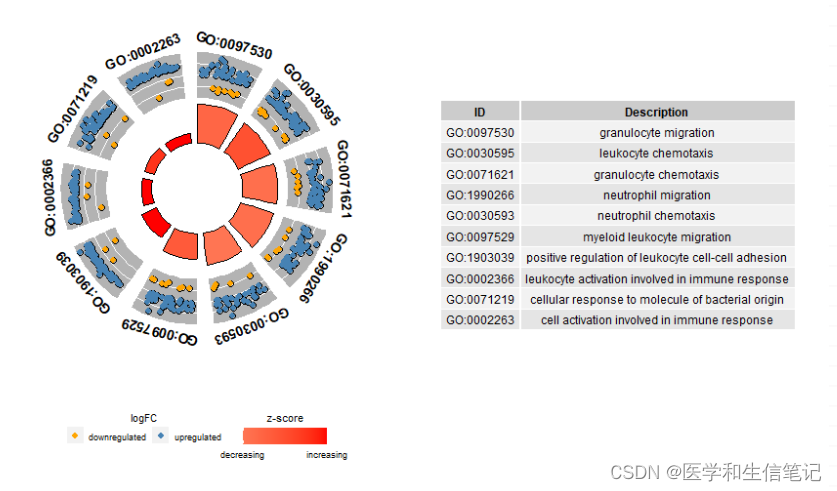
根据颜色映射应该就能看懂里面的圆环和外面的圆环分别表示什么了~
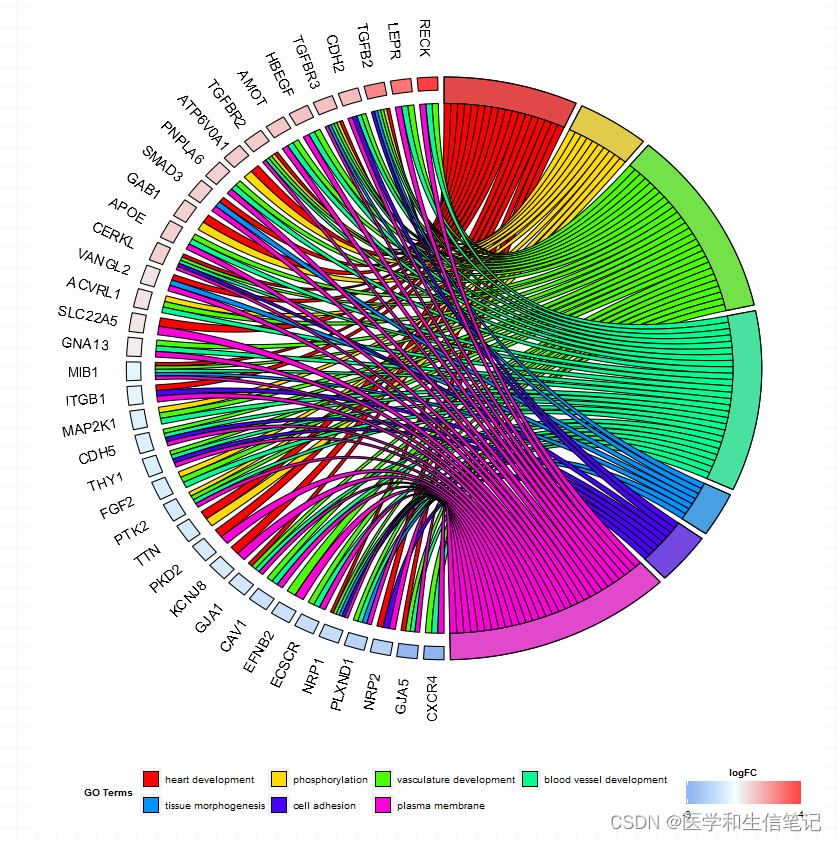
和弦图
前面是大致展示下富集分析结果的总体情况,和弦图可以展示特定条目和基因之间的关系。不过也需要提前整理好数据。
选择你感兴趣的基因和条目,继续添加到go_list这个列表中:
go_list$genes <- deg_limma[,c("ID","logFC")]
go_list$process <- c("plasma lipoprotein particle","lipoprotein particle",
"immunological synapse",
"platelet alpha granule lumen",
"protein complex involved in cell adhesion"
)
使用chord_dat函数继续添加数据:
chord <- chord_dat(circ, go_list$genes, go_list$process)
head(chord)
## plasma lipoprotein particle lipoprotein particle immunological synapse
## TIMP1 0 0 0
## SERPINA1 0 0 0
## VLDLR 1 1 0
## LIPC 1 1 0
## CTLA4 0 0 0
## LYN 0 0 0
## platelet alpha granule lumen protein complex involved in cell adhesion
## TIMP1 1 0
## SERPINA1 1 0
## VLDLR 0 0
## LIPC 0 0
## CTLA4 0 1
## LYN 0 1
## logFC
## TIMP1 2.125930
## SERPINA1 1.104712
## VLDLR -1.774415
## LIPC -1.606611
## CTLA4 2.806961
## LYN 1.214007
接下来画图就可以了:
GOChord(chord, space = 0.02,
gene.order = 'logFC',
gene.space = 0.25, #基因名和图形的距离
gene.size = 5, #基因名大小
nlfc = 1, # logfc列的数量?
lfc.col=c("red","white","blue"), # logfc颜色
lfc.min=-2, #对logfc标准化的最小值
lfc.max=2, #对logfc标准化的最大值
ribbon.col=c("#386CB0","#BEBADA","#D95F02","#8DD3C7","#8DA0CB"),#条目颜色
border.size=0.8, #条带宽度
process.label=8, #条目图例标签大小
limit=c(0,0)
)
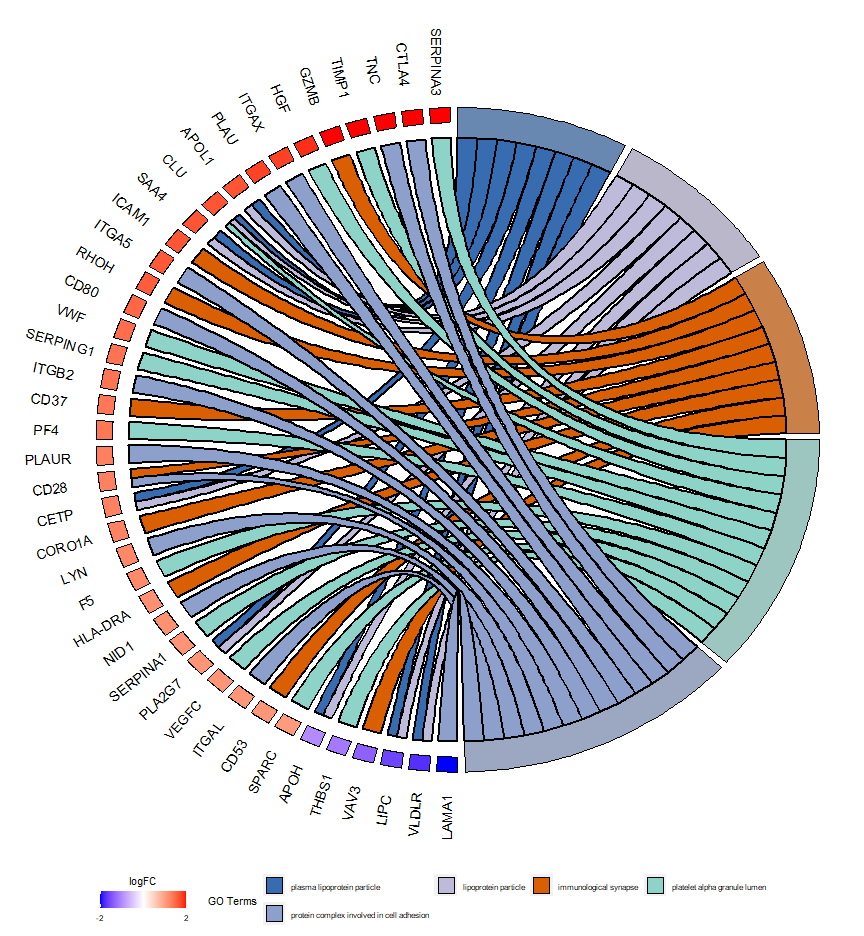
limit参数的两个数字意思:第一个数字:一个基因富集到最少几个条目上;第二个数字:一个条目最少有几个基因富集。
热图
在之前的enrichplot中介绍过了,这里就不多说了,一模一样。
GOHeat(chord,
nlfc = 1) # 1是用logfc,0是不用

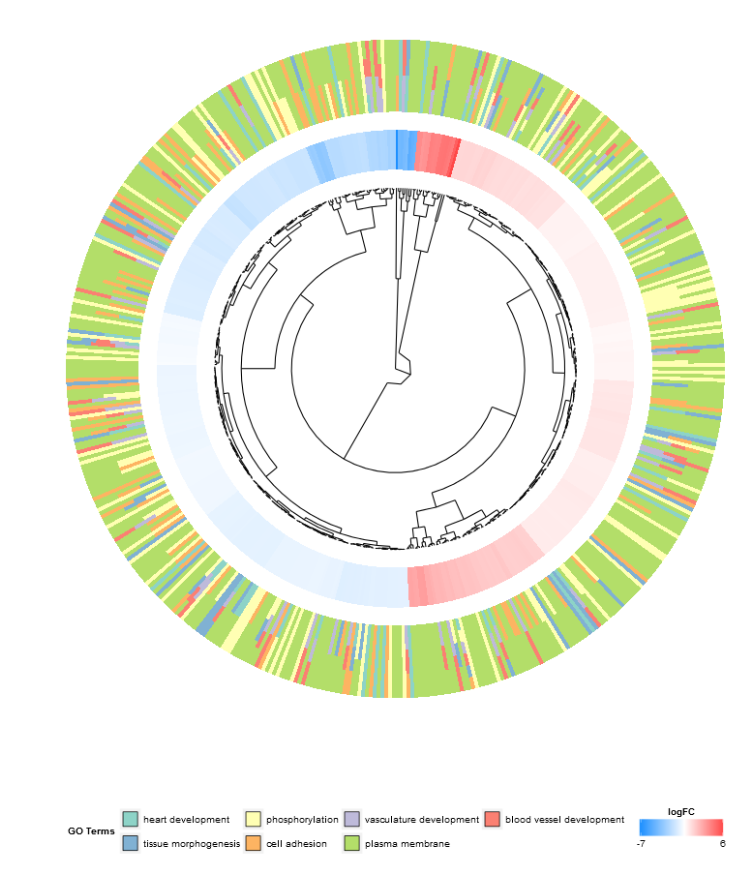
圆形聚类图
竟然也是用ggplot2画出来的,学习!
GOCluster(circ,
process = go_list$process, #感兴趣的条目
metric = "euclidean", # 聚类时计算距离的方法
clust = "average", # 聚类的方法
clust.by = 'logFC', # 聚类依据
term.width = 2, #条目圆环的宽度
lfc.width = 1, #logfc圆环的宽度
lfc.col = c('darkgoldenrod1', 'black', 'cyan1')
)
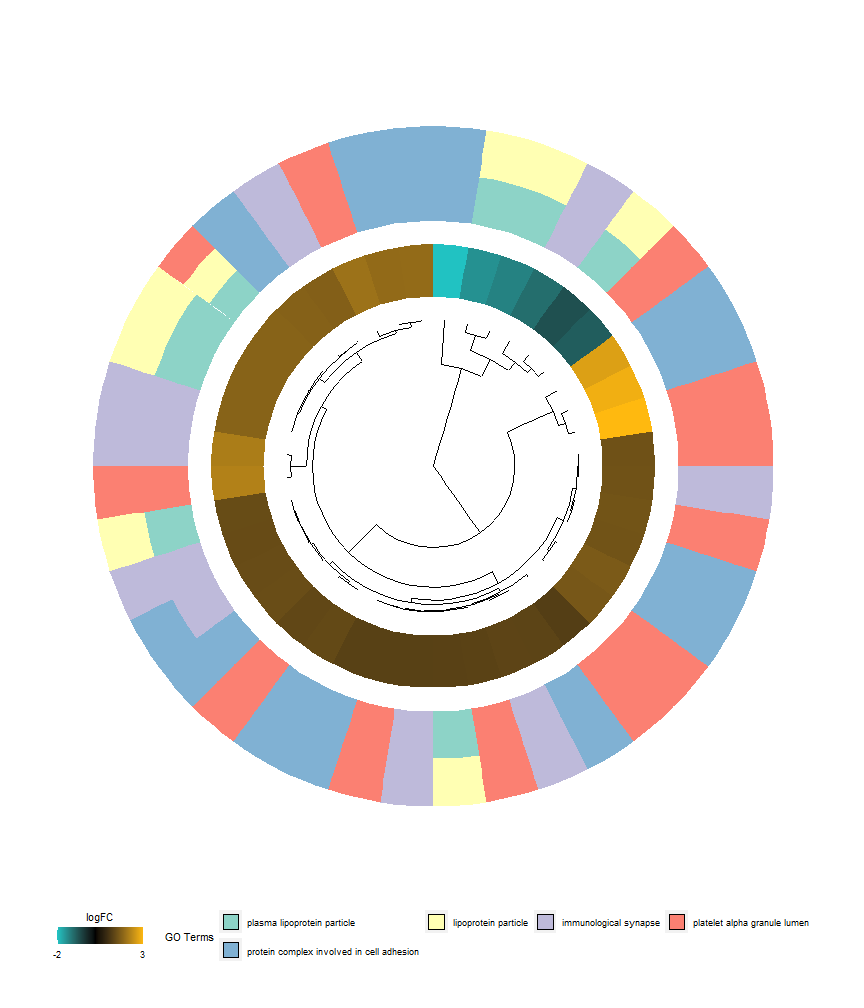
韦恩图
展示不同条目间重叠的基因,以及有几个基因富集到对应的条目上,最多支持3个条目。
l1 <- subset(circ, term == 'lipoprotein particle', c(genes,logFC))
l2 <- subset(circ, term == 'plasma lipoprotein particle', c(genes,logFC))
l3 <- subset(circ, term == 'immunological synapse', c(genes,logFC))
GOVenn(l1,l2,l3, label = c('lipoprotein particle',
'plasma lipoprotein particle',
'immunological synapse'))
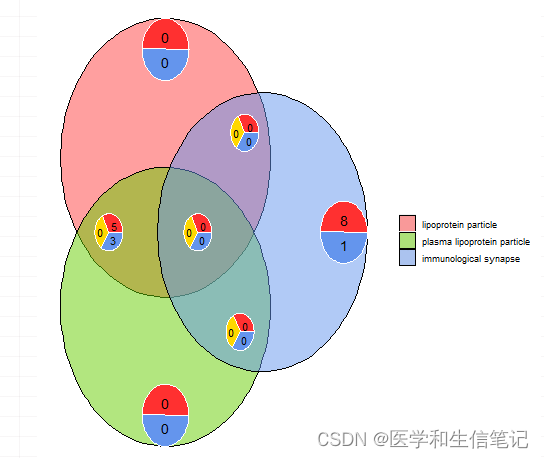
以上就是Goplot的演示,使用起来还是蛮简单的,准备数据的过程也并不是非常复杂,大家可以按照我的示例试一试,无非就是改几个列名,挑选几个自己感兴趣的条目。
后面会继续给大家带来富集分析可视化的内容。

















 992
992

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









