







 本文介绍如何从PATRIC数据库批量下载双歧杆菌基因组序列,并使用Python脚本进行序列下载。此外,还介绍了prokka基因注释工具和antiSMASH次级代谢产物分析工具的安装与使用,以及diamond序列对比工具的应用。
本文介绍如何从PATRIC数据库批量下载双歧杆菌基因组序列,并使用Python脚本进行序列下载。此外,还介绍了prokka基因注释工具和antiSMASH次级代谢产物分析工具的安装与使用,以及diamond序列对比工具的应用。
文章目录
双歧杆菌基因组序列批量下载
此模块介绍在PATRIC数据库中检索下载所有不同双歧杆菌菌株(大概1400株左右)的csv文件,再通过数据筛选和清洗得到我们将要下载的基因组序列信息。利用python脚本批量下载基因组序列(gbk文件或fasta文件)。
注:antiSMASH推荐使用gbk文件进行基因簇挖掘。
一、双歧杆菌基因组信息检索及下载
1.PATRIC数据库检索双歧杆菌(Bifidobacterium) 并下载序列信息csv文件
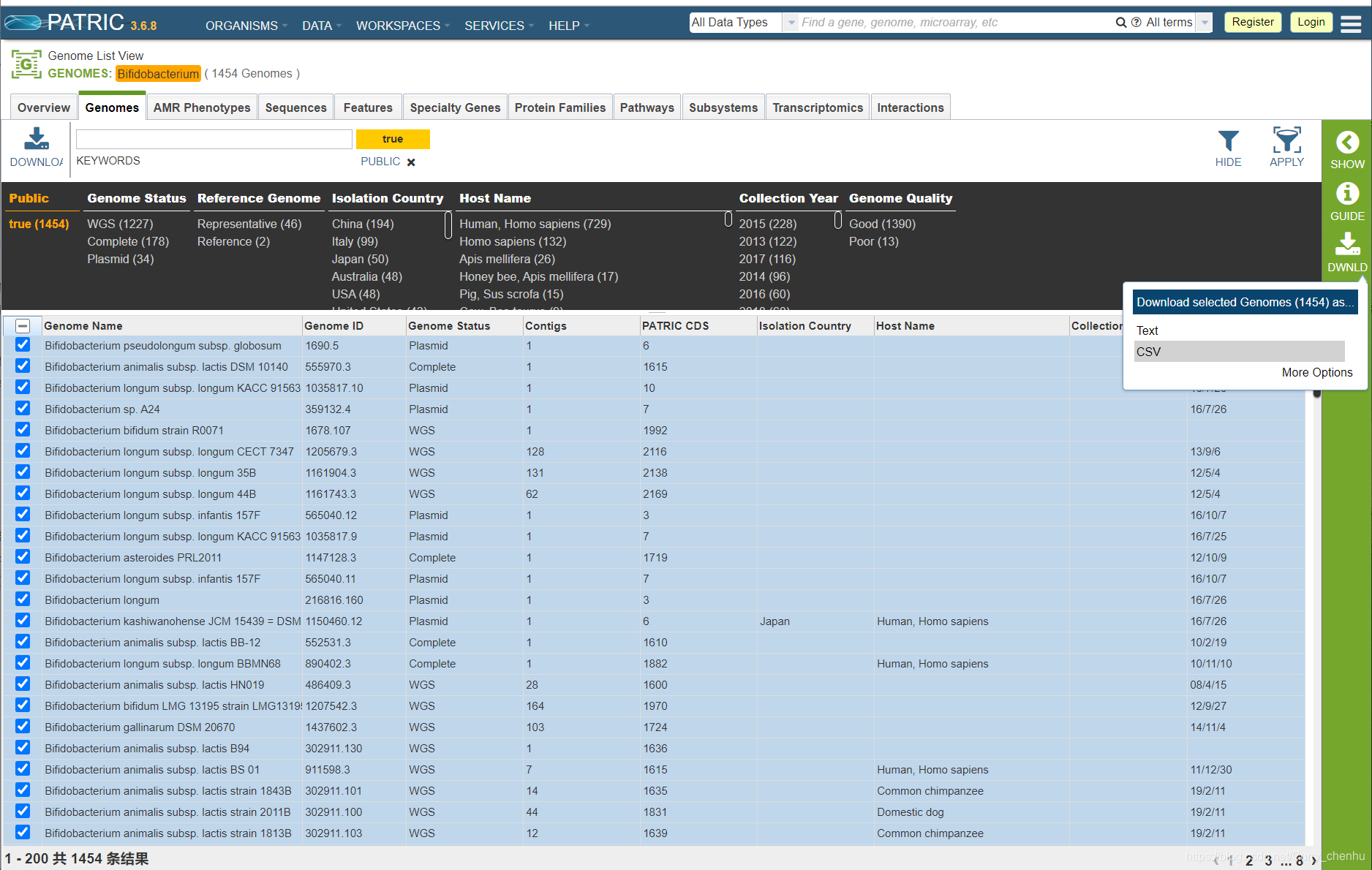
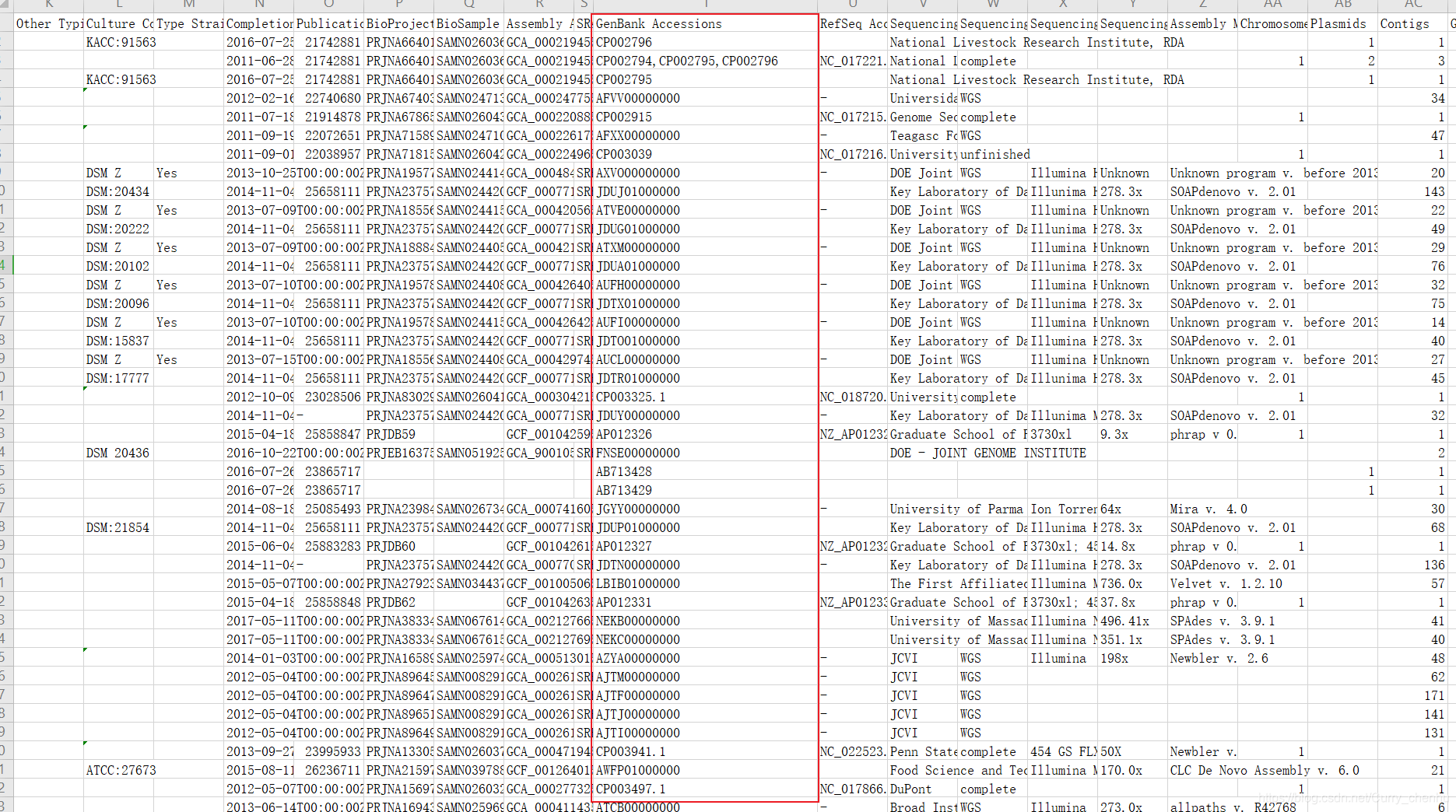
下载后的csv文件如下图所示,对这些序列信息进行筛选清洗,筛选出不重复的菌株序列。“GenBank Accessions”用于基因组序列下载(NCBI数据库)。
二、基因组序列批量下载
python脚本代码
from Bio import Entrez
import csv
# 参数设置
Entrez.email = "example@163.com"
Entrez.tool = "exampleScript"
csv_file = "complete2.csv" # csv文件路径
def get_gbk(csv_file):
"""
从csv文件中获取GenBank Accessions,返回基因组登记号列表(gbk_List)
:param csv_file: csv文件
:return: 含有所有GenBank Accessions的列表
"""
gbk_list =[] # 存储GenBank Accessions
# 读取csv文件
csv_flie = csv.reader(open(csv_file))
# 确定“GenBank Accessions”所在位置(即索引)
goal_index = 0
for l in csv_flie:
goal_index = l.index("GenBank Accessions")
break
# 读取所有组的信息找到对应的"GenBank Accessions"
for line in csv_flie:
gbk_list.append(line[goal_index])
return gbk_list
def download_gbk(gbk_list,goal_path):
"""
下载提供的基因组登记号列表中对应的genbank格式基因组序列,并存储到本地
:param gbk_list: "GenBank Accessions"列表
:param goal_path: gbk文件存储路径
:return: failed_list
"""
# 下载失败的基因组序列列表
failed_list = []
print("开始下载gbk格式基因组序列文件")
for gbk_id in gbk_list:
file_name = goal_path + gbk_id + ".gbk" # 指明路径:以GenBank Accessions命名文件
try:
handle = Entrez.efetch(db="nucleotide",id=gbk_id,rettype="gb",retmode="text")
file_handle=open(file_name,"w")
file_handle.write(handle.read())
file_handle.close()
# handle.close()
print("下载{}成功!".format(gbk_id))
except:
print("下载{}失败!!!".format(gbk_id))
failed_list.append(gbk_id)
return failed_list
def download_fasta(gbk_list,goal_path):
"""
下载提供的基因组登记号列表中对应的fasta格式基因组序列,并存储到本地
:param gbk_list: "GenBank Accessions"列表
:param goal_path: fasta文件存储路径
:return: failed_list
"""
# 下载失败的基因组序列列表
failed_list = []
print("开始下载fasta格式基因组序列文件")
for gbk_id in gbk_list:
file_name = goal_path + gbk_id + ".fasta" # 指明路径:以GenBank Accessions命名文件
try:
handle = Entrez.efetch(db="nucleotide", id=gbk_id, rettype="fasta", retmode="text")
file_handle = open(file_name, "w")
file_handle.write(handle.read())
file_handle.close()
# handle.close()
print("下载{}成功!".format(gbk_id),end="\t")
print(str(gbk_list.index(gbk_id)+1)+"/"+str(len(gbk_list)))
except:
print("下载{}失败!!!".format(gbk_id))
failed_list.append(gbk_id)
return failed_list
def try_again(failed_list,goal_path,rettype):
"""
对下载失败的基因组序列再尝试下载一次,返回两次都下载失败的对应基因组登记号列表
:param failed_list: 第一次下载失败的基因组序列列表
:param goal_path: 目标路径
:return: end_failed
"""
end_failed = []
for gbk_id in failed_list:
if rettype == "gb":
file_name = goal_path + gbk_id + ".gb" # 指明路径:以GenBank Accessions命名文件
else:
file_name = goal_path + gbk_id + "."+rettype
try:
handle = Entrez.efetch(db="nucleotide",id=gbk_id,rettype=rettype,retmode="text")
file_handle=open(file_name,"w")
file_handle.write(handle.read())
file_handle.close()
# handle.close()
print("第二次下载{}成功!".format(file_name))
except:
print("{}下载失败!!,请手动下载!".format(file_name))
end_failed.append(gbk_id)
return end_failed
def main():
# 获取基因组登记号列表
gbk_list=get_gbk(csv_file)
gbk_list = []
with open("results.csv","r") as f:
f_csv = csv.reader(f)
for row in f_csv:
gbk_list.append(row[1])
# 下载对应gbk格式基因组序列文件,并打印下载失败的登记号
failed_gbkdown=download_gbk(gbk_list,".\\gbk_file\\")
failed_gbklist=try_again(failed_gbkdown,".\\gbk_file\\","gb")
print("需手动下载的基因组序列(gb格式):{}".format(failed_gbklist))
# 下载对应fasta格式基因组序列文件,并打印下载失败的登记号 需要选择数据库protein或nucleotide
failed_fastadown = download_fasta(gbk_list,".\\fasta_file\\")
failed_fastalist = try_again(failed_fastadown,".\\fasta_file\\","fasta")
print("需手动下载的基因组序列(fasta格式):{}".format(failed_fastalist))
if __name__ == '__main__':
main()
需要自己新建两个存储基因组序列的文件夹(gbk_file、fasta_file)。若你检索的是protein数据库,将代码中三个函数中的db=“nucleotide”,改为“protein”即可。genbank登录号不一定需要来自csv文件中,如果有基因组登录号列表,可注释main函数中的 gbk_list=get_gbk(csv_file),将下载你提供的登录号列表基因组。
prokka基因组注释
直接从下载gbk序列文件,可能注释不全或者未注释,导致antismash无结果。索引统一对下载下来的fasta文件进行prokka基因注释再进行antismash分析。
一、prokka的安装
参考文档:官方安装帮助文档,基因注释Prokka
可能会出现perl的依赖包缺少的情况,可以参考我前面发过的文章。
二、prokka使用
输入文件类型:fasta
prokka final.contigs.fa --outdir prokka_annotation --prefix metagG --metagenome --kingdom Bacteria
具体参数可参考官方文档
三、结果
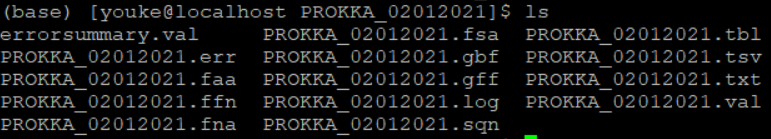
prokka注释结果将会产生如下不同的文件,其中后缀gbf可改为gbk,即为antismash输入文件。
antiSMASH安装及使用
一、antiSMASH简介
antiSMASH可以对细菌和真菌基因组中的次级代谢产物生物合成基因簇进行全基因组范围内的快速鉴定,注释和分析。它与早先已发布的大量计算机辅助二级代谢物分析工具集成和交叉链接。
antiSMASH网页版
二、antiSMASH安装与使用
1.安装
我的安装环境是Centos服务器。具体的安装过程我前面的文章有介绍到,这里就不再赘述了。antiSMASH安装与使用
在安装依赖库的时候注意各个依赖库的版本,版本尽量选择antiSMASH官方测试过的版本(不然痛苦面具说出就出…)。参考官方文档

2.使用
输入文件类型:gbk
antismash *.gbk 就够用了,更多参数可以查看帮助文档 antismash -h
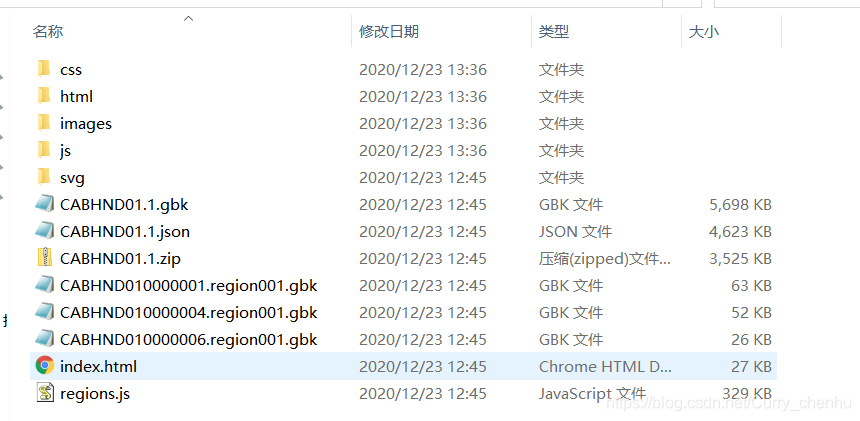
结果
结果生成一个html文件夹,可以点击index.html查看结果
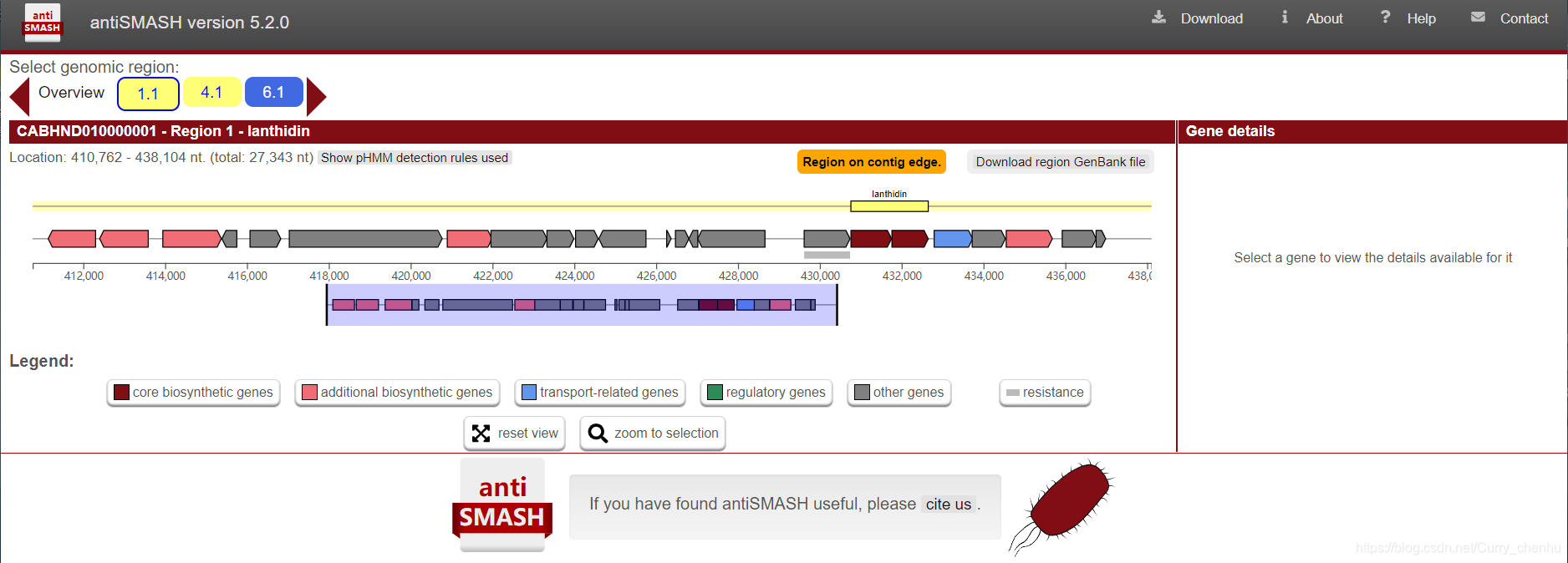
更多结果解读参考官方手册
核心基因的同源性比较
序列对比提前安装好blast或diamond软件(这里介绍的是diamond),并下载nr数据库。
一、提取核心基因AA序列
利用python中的selenium库提取序列,源代码
二、diamond序列对比工具
1.简介
DIAMOND是用于蛋白质和翻译的DNA搜索的序列比对器,旨在用于大序列数据的高性能分析。据分析,当针对NCBI-nr数据库进行显着比对,预期值低于10 -3时,DIAMOND比BLAST比对大约快20,000倍,并具两个工具有相似的灵敏度水平。
2.diamond的安装
wget http://github.com/bbuchfink/diamond/releases/download/v0.9.24/diamond-linux64.tar.gz
tar xzf diamond-linux64.tar.gz
# 将diamond添加到环境变量中去(~/.bashrc)
数据库下载(nr数据库)
wget ftp://ftp.ncbi.nlm.nih.gov/blast/db/FASTA/nr.gz
数据库有点大,可以结合nohup和&后台下载
nohup wget ftp://ftp.ncbi.nlm.nih.gov/blast/db/FASTA/nr.gz >> nohup.out 2>&1 &
数据库下载完后,开始建库 diamond makedb --in nr.fa -d nr ,建库结束后就可以开始diamond的运行了。
3.diamond的使用
diamond blastp -d nr -q reads.fasta -f 6 -o reads.blast -k 10
-d 前面所建的nr数据库
-q 待比对的序列
-f 输出文件格式
-k 设置每个query比对结果的最大匹配数目
更多参数解读参考文章
结果
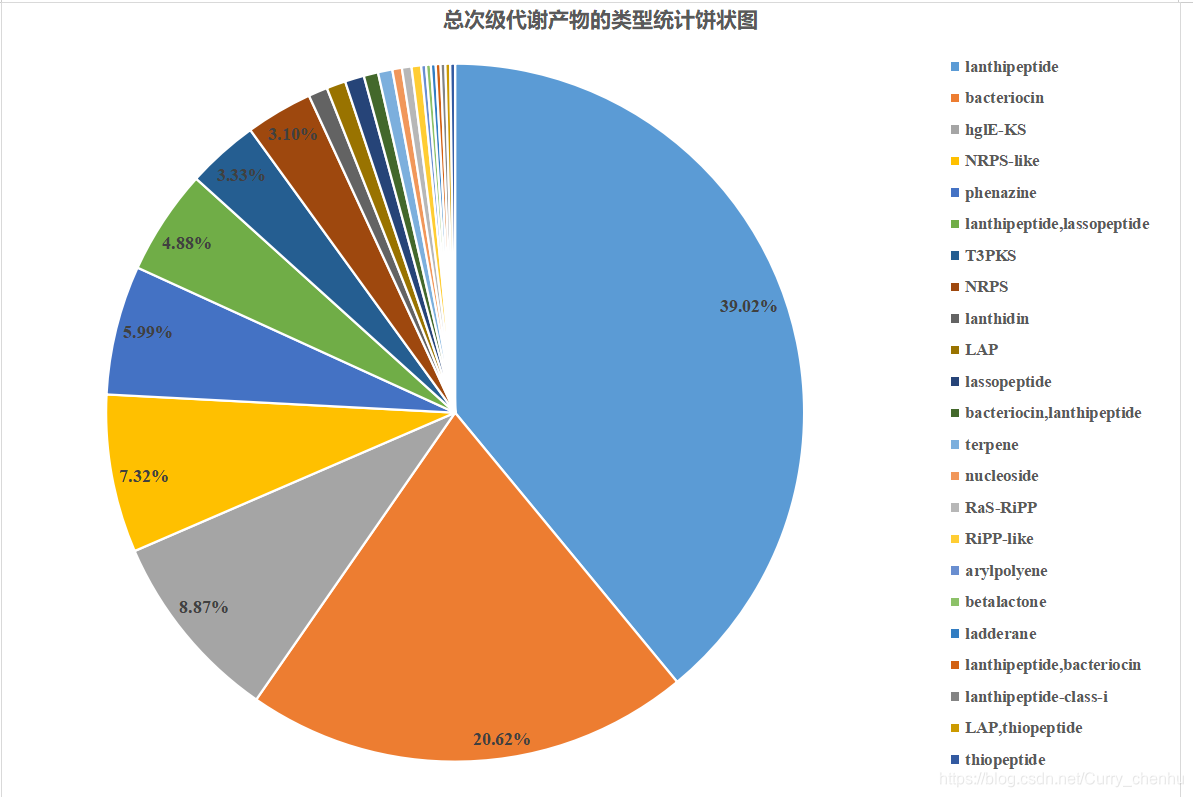
1.次级代谢产物类型在双歧杆菌中的分布
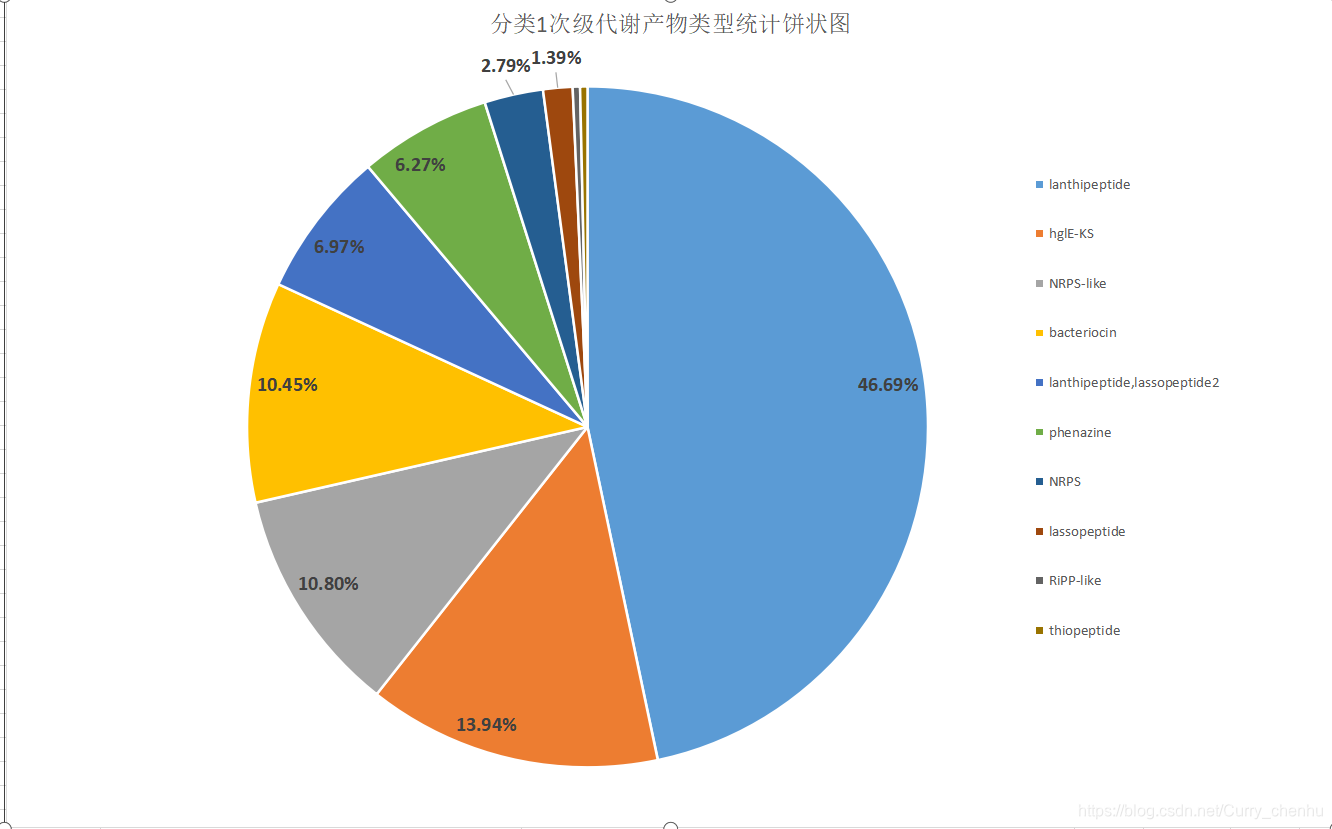
2.对同源性比较结果进行分类,得知哪一些次级代谢产物是双歧杆菌特有的,还可以挖掘到一些未发布或者没有研究过的双歧杆菌次级代谢产物类型。
码字不易,转载请标注出处。

















 1107
1107

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









