









摘要
模拟相关文件:
cpt: 检查点文件,保存模拟状态,可用于恢复模拟。
edr/ene: 能量文件,存储能量数据,可使用 gmx energy 分析。
trr: 模拟轨迹文件,记录坐标、速度、力等。
tpr: 运行输入文件,包含分子拓扑、模拟参数等,二进制格式。
结构与拓扑文件:
gro: 分子结构文件,包含分子坐标和速度,支持简单的轨迹操作。
pdb: 蛋白质数据库文件,描述分子结构,可包含多个模型。
itp/top: 拓扑文件,定义分子及力场参数。
rtp: 残基拓扑文件,用于生成蛋白质拓扑。
索引与分组文件:
ndx: 索引文件,定义原子或分子的分组,便于分析。
n2t/r2b: 原子与残基名称的翻译文件,支持不同力场或命名系统间的转换。
输入参数文件:
mdp: 模拟参数文件,控制分子动力学模拟。
hdb: 氢数据库文件,生成或修复缺失的氢原子。
tdb:端基数据库文件,定义多肽链的末端残基。
分析与可视化文件:
eps/m2p: 用于生成 PostScript 格式的可视化图形文件。
xpm: 矩阵文件,可通过 gmx xpm2ps 转换为图像。
通用文件:
dat/out: 通用输入或输出文件,无固定格式。
log: 日志文件,记录程序运行信息。
mtx: 矩阵文件,用于保存 Hessian
极为稀少 快收藏吧
GROMACS的文件格式说明
atp文件
atp文件包含有关原子类型的一般信息,如原子序数和以原子质量为单位的质量。
arn文件
arn文件允许原子从它们的力场名称重命名为IUPAC/PDB定义的名称,以便更容易地可视化和识别。
cpt 文件
cpt 文件扩展名代表便携式检查点文件。模拟的完整状态存储在检查点文件中,包括扩展恒温器/恒压器变量、随机数状态和 NMR 时间平均数据。域分解还存储了一些分解设置信息。
参阅gmx mdrun。.
dat 文件
带有 dat 文件扩展名的文件包含通用输入或输出。由于无法对所有数据文件格式进行分类,GROMACS 有一种称为 dat 的通用文件格式,但未给出其格式。
edi 文件
带有 edi 文件扩展名的文件包含有关gmx mdrun的信息 ,用于运行具有基本动力学约束的分子动力学。以前可以通过WHAT IF程序中提供的选项生成这些信息。
edr 文件
edr 文件扩展名代表便携式能量文件。能量使用 xdr 协议存储。
可请参阅gmx 能量。
ene 文件
ene 文件扩展名代表二进制能量文件。它保存着gmx mdrun期间生成的能量。
该文件可以使用程序 gmx eneconv转换为可移植能量文件(可跨硬件平台移植),即edr文件。
eps 文件
eps 文件格式不是特殊的 GROMACS 格式,而只是标准 PostScript™ 的变体。下面包含由gmx xpm2ps程序生成的 eps 文件示例。它显示了肽的二级结构随时间的变化。
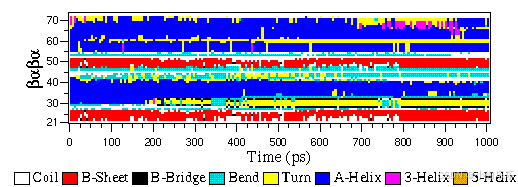
g96 文件
扩展名为 g96 的文件可以是 GROMOS-96 初始/最终配置文件或坐标轨迹文件,也可以是两者的组合。该文件是固定格式,所有浮点数都写为 15.9(文件可能会变得很大)。GROMACS 按给定顺序支持以下数据块:
- 标题块:
- TITLE(强制的)
- 框架块:
-
TIMESTEP(选修的)
-
POSITION/POSITIONRED(强制的)
-
VELOCITY/VELOCITYRED(选修的)
-
BOX(选修的)
-
请注意,所有 GROMACS 程序都可以读取压缩文件或 g-zipped 文件。
gro 文件
带有 gro 文件扩展名的文件包含 Gromos87 格式的分子结构。只需将文件连接起来,gro 文件即可用作轨迹。将尝试从每帧的标题字符串中读取时间值,该时间值前面应加上“ t=”,如下面的示例所示。
下面附有一个样本:
MD of 2 waters, t= 0.0
6
1WATER OW1 1 0.126 1.624 1.679 0.1227 -0.0580 0.0434
1WATER HW2 2 0.190 1.661 1.747 0.8085 0.3191 -0.7791
1WATER HW3 3 0.177 1.568 1.613 -0.9045 -2.6469 1.3180
2WATER OW1 4 1.275 0.053 0.622 0.2519 0.3140 -0.1734
2WATER HW2 5 1.337 0.002 0.680 -1.0641 -1.1349 0.0257
2WATER HW3 6 1.326 0.120 0.568 1.9427 -0.8216 -0.0244
1.82060 1.82060 1.82060
各行包含以下信息(从上到下):
- 标题字符串(自由格式字符串,ps 中’ t='后的可选时间)
- 原子数(自由格式整数)
- 每个原子一行(固定格式,见下文)
- 盒子向量(自由格式,空格分隔的实数),值:v1(x) v2(y) v3(z) v1(y) v1(z) v2(x) v2(z) v3(x)
v3(y),最后 6 个值可以省略(它们将被设置为零)。GROMACS 仅支持 v1(y)=v1(z)=v2(z)=0 的盒子。
此格式是固定的,即所有列都处于固定位置。可选(目前仅限使用 trjconv)您可以编写具有任意小数位数的 gro 文件,格式将为带有 n+5小数位的位置n(n+1 对于速度)而不是8带有3(对于 4速度)。读取后,精度将从小数点之间的距离推断出来(将是 n+5)。列包含以下信息(从左到右):
残基数(5 个位置,整数)
残基名称(5个字符)
原子名称(5 个字符)
原子数(5 位,整数)
位置(单位为 nm,xyz 分为 3 列,每列 8 个位置,有 3 位小数)
速度(单位为 nm/ps(或 km/s),xyz 分为 3 列,每列 8 位,有 4 位小数)
请注意,单独的分子或离子(例如水或 Cl-)被视为残基。如果您想在不使用 GROMACS 库的情况下在自己的程序中编写此类文件,则可以使用以下格式:
C format
“%5d%-5s%5s%5d%8.3f%8.3f%8.3f%8.4f%8.4f%8.4f”
Fortran format
(i5,2a5,i5,3f8.3,3f8.4)
请注意,这是书写的格式,因为在上面的例子中,字段可能没有空格,因此不能用 C 中的相同格式语句读取。
hdb 文件
hdb文件扩展名表示氢数据库,gmx pdb2gmx在构建最初缺失或使用-ignh删除的氢原子时需要这样的文件。
itp 文件
itp文件扩展名代表包含拓扑。这些文件包含在拓扑文件中(扩展名为top)。
log文件
日志文件是由一些GROMACS程序生成的,通常是人类可读的格式。使用更多的日志文件。
m2p文件
m2p文件格式包含gmx xpm2ps程序的输入选项。
当您查看gmx xpm2ps的PostScript(tm)输出时,所有这些选项都非常容易理解。
; Command line options of xpm2ps override the parameters in this file
black&white = no ; Obsolete
titlefont = Times-Roman ; A PostScript Font
titlefontsize = 20 ; Font size (pt)
legend = yes ; Show the legend
legendfont = Times-Roman ; A PostScript Font
legendlabel = ; Used when there is none in the .xpm
legend2label = ; Used when merging two xpm's
legendfontsize = 14 ; Font size (pt)
xbox = 2.0 ; x-size of a matrix element
ybox = 2.0 ; y-size of a matrix element
matrixspacing = 20.0 ; Space between 2 matrices
xoffset = 0.0 ; Between matrix and bounding box
yoffset = 0.0 ; Between matrix and bounding box
x-major = 20 ; Major ticks on x axis every .. frames
x-minor = 5 ; Id. Minor ticks
x-firstmajor = 0 ; First frame for major tick
x-majorat0 = no ; Major tick at first frame
x-majorticklen = 8.0 ; x-majorticklength
x-minorticklen = 4.0 ; x-minorticklength
x-label = ; Used when there is none in the .xpm
x-fontsize = 16 ; Font size (pt)
x-font = Times-Roman ; A PostScript Font
x-tickfontsize = 10 ; Font size (pt)
x-tickfont = Helvetica ; A PostScript Font
y-major = 20
y-minor = 5
y-firstmajor = 0
y-majorat0 = no
y-majorticklen = 8.0
y-minorticklen = 4.0
y-label =
y-fontsize = 16
y-font = Times-Roman
y-tickfontsize = 10
y-tickfont = Helvetica
mdp 文件
下面是一个示例 mdp 文件。项目的顺序并不重要,但如果您输入两次相同的内容,则使用最后一个( gmx grompp在覆盖值时会给您一个注释)。左侧的破折号和下划线将被忽略。
选项的值是水中蛋白质 1 纳秒 MD 运行的值。
注意:所选的参数(例如,短程截止)取决于所使用的力场。
integrator = md
dt = 0.002
nsteps = 500000
nstlog = 5000
nstenergy = 5000
nstxout-compressed = 5000
continuation = yes
constraints = all-bonds
constraint-algorithm = lincs
cutoff-scheme = Verlet
coulombtype = PME
rcoulomb = 1.0
vdwtype = Cut-off
rvdw = 1.0
DispCorr = EnerPres
tcoupl = V-rescale
tc-grps = Protein SOL
tau-t = 0.1 0.1
ref-t = 300 300
pcoupl = Parrinello-Rahman
tau-p = 2.0
compressibility = 4.5e-5
ref-p = 1.0
通过此输入,gmx grompp将生成一个带有默认名称的注释文件 mdout.mdp。该文件将包含上述选项以及未明确设置的所有其他选项,并显示其默认值。
mtx文件
扩展名为 mtx 的文件包含一个矩阵。
该文件格式与trr格式相同。目前,此文件格式仅用于 hessian 矩阵,这些矩阵由gmx mdrun生成,并由 gmx nmeig读取。
ndx 文件
GROMACS 索引文件(通常称为 index.ndx)包含一些用户可定义的原子集。
大多数分析程序和预处理器(gmx grompp)都可以读取该文件。当没有提供索引文件时,大多数这些程序都会创建默认索引组,因此只有在需要特殊组时才需要创建索引文件。
首先,组名写在方括号内。以下原子编号可以分散到任意多行。原子编号从 1 开始。
示例文件如下:
[ Oxygen ]
1 4 7
[ Hydrogen ]
2 3 5 6
8 9
有两组,总共九个原子。第一组 氧有3个元素。第二组氢有6个元素。
可用的索引文件生成工具是: gmx make_ndx。
n2t 文件
此 GROMACS 文件可用于执行结构文件中的原子名称与相应原子类型之间的基本转换。
这在使用gmx x2top等实用程序时非常有用,但用户应注意,此文件中的知识极其有限。
示例文件(share/top/gromos53a5.ff/atomname2type.n2t)在此处:
H H 0.408 1.008 1 O 0.1
O OA -0.674 15.9994 2 C 0.14 H 0.1
C CH3 0.000 15.035 1 C 0.15
C CH0 0.266 12.011 4 C 0.15 C 0.15 C 0.15 O 0.14
文件格式的简短描述如下:
第 1 列:原子的元素符号/原子名称中的第一个字符。
第 2 列:要分配的原子类型。
第 3 列:要分配的费用。
第 4 列:原子的质量。
第 5 列:与该原子键合的其他原子的数量 N。后面的字段数量与此数字相关;对于每个原子,都有一个元素符号和其键长的参考距离。
第 6 列及以后:元素符号和 N 连接(第 5 列)与被分配参数的原子(第 1 列)的参考键长。参考键长与本文件中指定的值有 +/- 10% 的误差。任何超出此误差的键都不会被识别为与被分配参数的原子相连。
out 文件
带有 out 文件扩展名的文件包含通用输出。
由于无法对所有数据文件格式进行分类,GROMACS 有一种通用文件格式,称为 out,但未给出任何格式。
pdb 文件
扩展名为pdb的文件是蛋白质数据库文件格式的分子结构文件。
蛋白质数据库文件格式描述了分子结构中原子的位置。从ATOM和HETATM记录中读取坐标,直到文件结束或遇到ENDMDL记录。GROMACS程序可以在CRYST1条目中读写模拟框。pdb格式也可以用作轨迹格式:可以从一个文件中读取或写入由ENDMDL分隔的多个结构。
例子
pdb 文件应如下所示:
ATOM 1 H1 LYS 1 14.260 6.590 34.480 1.00 0.00
ATOM 2 H2 LYS 1 13.760 5.000 34.340 1.00 0.00
ATOM 3 N LYS 1 14.090 5.850 33.800 1.00 0.00
ATOM 4 H3 LYS 1 14.920 5.560 33.270 1.00 0.00
...
...
rtp 文件
rtp 文件扩展名代表残基拓扑。gmx pdb2gmx需要这样的文件来为pdb文件 中的蛋白质创建 GROMACS 拓扑 。该文件包含 4 种键合相互作用和残基条目的默认相互作用类型,这些条目由原子和可选的键、角二面角和不正确的分子组成。可以为键、角、二面角和不正确的分子添加参数,这些参数将覆盖itp文件中的标准参数。这只应用于特殊情况。可以为每个键合相互作用添加一个字符串来代替参数,该字符串将复制到顶部文件,这用于 GROMOS96 力场。
gmx pdb2gmx自动生成所有角度,这意味着该[angles]字段仅用于覆盖itp参数。
gmx pdb2gmx自动为每个可旋转键生成一个适当的二面角,最好是在重原子上。当[dihedrals]使用该字段时,不会为与指定二面角相对应的键生成其他二面角。可以在可旋转键上放置多个二面角。
gmx pdb2gmx将排除数设置为 3,这意味着最多排除由 3 个键连接的原子之间的相互作用。所有由 3 个键分隔的原子对(氢键对除外)都会生成对相互作用。当需要排除更多相互作用或不应生成某些对相互作用时,[exclusions]可以添加一个字段,后跟单独行上的原子名称对。这些原子之间的所有非键合和对相互作用都将被排除。
下面附有一个示例。
[ bondedtypes ] ; mandatory
; bonds angles dihedrals impropers
1 1 1 2 ; mandatory
[ GLY ] ; mandatory
[ atoms ] ; mandatory
; name type charge chargegroup
N N -0.280 0
H H 0.280 0
CA CH2 0.000 1
C C 0.380 2
O O -0.380 2
[ bonds ] ; optional
;atom1 atom2 b0 kb
N H
N CA
CA C
C O
-C N
[ exclusions ] ; optional
;atom1 atom2
[ angles ] ; optional
;atom1 atom2 atom3 th0 cth
[ dihedrals ] ; optional
;atom1 atom2 atom3 atom4 phi0 cp mult
[ impropers ] ; optional
;atom1 atom2 atom3 atom4 q0 cq
N -C CA H
-C -CA N -O
[ ZN ]
[ atoms ]
ZN ZN 2.000 0
r2b 文件
r2b文件将残基名称翻译为在不同力场中具有不同名称的残基,或根据其质子化状态具有不同名称的残基。
tdb 文件
TDB文件包含有关氨基酸末端的信息,该末端可以放置在多肽链的末端。
tex 文件
我们用LaTeX来处理文档。虽然输入不太方便,但它比文字处理器有一些优势。
LaTeX对格式化非常了解,可能比你了解得多。
输入很清晰,你总是知道自己在做什么
它可以制作从信件到论文的任何内容
更多内容…
tng文件
文件扩展名为.tng的文件可以包含与模拟轨迹相关的所有类型的数据。例如,它可能包含坐标、速度、力和/或能量。各种mdp文件选项控制gmx mdrun写入哪些文件,是否使用压缩方式写入数据,以及压缩的有损程度。这个文件是可移植的二进制格式,可以用gmx dump读取。
gmx dump -f traj.tng
如果你的阅读速度不是那么快:
gmx dump -f traj.tng | less
您还可以使用以下方法快速查看文件内容(帧数等):
gmx check -f traj.tng
top 文件
top 文件扩展名代表拓扑。它是一个 ascii 文件,由gmx grompp读取并处理它,然后创建二进制拓扑(tpr文件)。
下面附有一个示例文件:
;
; Example topology file
;
[ defaults ]
; nbfunc comb-rule gen-pairs fudgeLJ fudgeQQ
1 1 no 1.0 1.0
; The force field files to be included
#include "rt41c5.itp"
[ moleculetype ]
; name nrexcl
Urea 3
[ atoms ]
; nr type resnr residu atom cgnr charge
1 C 1 UREA C1 1 0.683
2 O 1 UREA O2 1 -0.683
3 NT 1 UREA N3 2 -0.622
4 H 1 UREA H4 2 0.346
5 H 1 UREA H5 2 0.276
6 NT 1 UREA N6 3 -0.622
7 H 1 UREA H7 3 0.346
8 H 1 UREA H8 3 0.276
[ bonds ]
; ai aj funct c0 c1
3 4 1 1.000000e-01 3.744680e+05
3 5 1 1.000000e-01 3.744680e+05
6 7 1 1.000000e-01 3.744680e+05
6 8 1 1.000000e-01 3.744680e+05
1 2 1 1.230000e-01 5.020800e+05
1 3 1 1.330000e-01 3.765600e+05
1 6 1 1.330000e-01 3.765600e+05
[ pairs ]
; ai aj funct c0 c1
2 4 1 0.000000e+00 0.000000e+00
2 5 1 0.000000e+00 0.000000e+00
2 7 1 0.000000e+00 0.000000e+00
2 8 1 0.000000e+00 0.000000e+00
3 7 1 0.000000e+00 0.000000e+00
3 8 1 0.000000e+00 0.000000e+00
4 6 1 0.000000e+00 0.000000e+00
5 6 1 0.000000e+00 0.000000e+00
[ angles ]
; ai aj ak funct c0 c1
1 3 4 1 1.200000e+02 2.928800e+02
1 3 5 1 1.200000e+02 2.928800e+02
4 3 5 1 1.200000e+02 3.347200e+02
1 6 7 1 1.200000e+02 2.928800e+02
1 6 8 1 1.200000e+02 2.928800e+02
7 6 8 1 1.200000e+02 3.347200e+02
2 1 3 1 1.215000e+02 5.020800e+02
2 1 6 1 1.215000e+02 5.020800e+02
3 1 6 1 1.170000e+02 5.020800e+02
[ dihedrals ]
; ai aj ak al funct c0 c1 c2
2 1 3 4 1 1.800000e+02 3.347200e+01 2.000000e+00
6 1 3 4 1 1.800000e+02 3.347200e+01 2.000000e+00
2 1 3 5 1 1.800000e+02 3.347200e+01 2.000000e+00
6 1 3 5 1 1.800000e+02 3.347200e+01 2.000000e+00
2 1 6 7 1 1.800000e+02 3.347200e+01 2.000000e+00
3 1 6 7 1 1.800000e+02 3.347200e+01 2.000000e+00
2 1 6 8 1 1.800000e+02 3.347200e+01 2.000000e+00
3 1 6 8 1 1.800000e+02 3.347200e+01 2.000000e+00
[ dihedrals ]
; ai aj ak al funct c0 c1
3 4 5 1 2 0.000000e+00 1.673600e+02
6 7 8 1 2 0.000000e+00 1.673600e+02
1 3 6 2 2 0.000000e+00 1.673600e+02
; Include SPC water topology
#include "spc.itp"
[ system ]
Urea in Water
[ molecules ]
Urea 1
SOL 1000
tpr 文件
tpr 文件扩展名代表可移植二进制运行输入文件。此文件包含模拟的起始结构、分子拓扑和所有模拟参数。由于此文件为二进制格式,因此无法使用普通编辑器读取。要读取可移植二进制运行输入文件,请输入:
gmx dump -s topol.tpr
您还可以使用以下方法比较两个 tpr 文件:
gmx check -s1 top1 -s2 top2 | less
trr 文件
文件扩展名为trr的文件包含模拟的轨迹。在这个文件中,所有的坐标、速度、力和能量都按照您在mdp文件中告诉GROMACS的那样打印出来。这个文件是可移植的二进制格式,可以用gmx dump读取:
gmx dump -f traj.trr
如果你的阅读速度不是那么快:
gmx dump -f traj.trr | less
您还可以使用以下方法快速查看文件内容(帧数等):
% gmx check -f traj.trr
vsd文件
vsd 文件包含有关如何在力场中将虚拟位点放置在多个不同分子上的信息。
xdr文件
GROMACS 使用 XDR 文件格式在内部存储坐标文件之类的内容。
xpm
GROMACS xpm 文件格式与 XPixMap 格式兼容,用于存储矩阵数据。因此,可以使用 XV 等程序直接查看 GROMACS xpm 文件。或者,可以将它们导入 GIMP 并缩放到 300 DPI,对字体和图形使用强抗锯齿。xpm 文件中的第一个矩阵数据行对应于最后一个矩阵行。除了 XPixMap 格式外,GROMACS xpm 文件可能包含额外字段。在使用 gmx xpm2ps将 xpm 文件转换为 EPS 时,将使用这些字段中的信息。可选的额外字段包括:
- 在gv_xpm声明之前:标题、图例、x-label、y-label和类型,后面都跟一个字符串。图例字段决定图例标题。类型字段必须后跟“连续”或“离散”,这决定了将在EPS文件中绘制哪种类型的图例,默认类型是连续的。
- xpm颜色映射项后面可以跟一个字符串,该字符串是该颜色的标签。
- 在颜色图和矩阵数据之间,字段x轴或y轴可能会出现,然后是该轴的打勾标记。
下面的示例GROMACS xpm文件包含所有额外的字段。额外字段中的c注释分隔符和冒号是可选的。
/* XPM */
/* This matrix is generated by gmx rms. */
/* title: "Backbone RMSD matrix" */
/* legend: "RMSD (nm)" */
/* x-label: "Time (ps)" */
/* y-label: "Time (ps)" */
/* type: "Continuous" */
static char * gv_xpm[] = {
"13 13 6 1",
"A c #FFFFFF " /* "0" */,
"B c #CCCCCC " /* "0.0399" */,
"C c #999999 " /* "0.0798" */,
"D c #666666 " /* "0.12" */,
"E c #333333 " /* "0.16" */,
"F c #000000 " /* "0.2" */,
/* x-axis: 0 40 80 120 160 200 240 280 320 360 400 440 480 */
/* y-axis: 0 40 80 120 160 200 240 280 320 360 400 440 480 */
"FEDDDDCCCCCBA",
"FEDDDCCCCBBAB",
"FEDDDCCCCBABC",
"FDDDDCCCCABBC",
"EDDCCCCBACCCC",
"EDCCCCBABCCCC",
"EDCCCBABCCCCC",
"EDCCBABCCCCCD",
"EDCCABCCCDDDD",
"ECCACCCCCDDDD",
"ECACCCCCDDDDD",
"DACCDDDDDDEEE",
"ADEEEEEEEFFFF"
xtc 文件
xtc 格式是一种可移植的轨迹格式。它使用为 Unix NFS 系统创建的xdr例程来写入和读取数据。轨迹使用精度较低的算法写入,其工作方式如下:将坐标(以 nm 为单位)乘以比例因子(通常为 1000),这样就得到以 pm 为单位的坐标。这些坐标被四舍五入为整数值。然后执行其他几个技巧,例如利用原子在序列中靠近的事实,原子在空间中通常也靠近(例如水分子)。为此,xdr库扩展了一个特殊例程来写入 3-D 浮点坐标。该例程最初由 Frans van Hoesel 作为 Europort 项目的一部分编写。可以通过此链接获取其更新版本。
所有数据均通过调用xdr例程进行存储。
int magic
一个神奇的数字,对于当前文件版本,它的值是1995。
int natoms
轨道上原子的数目。
int step
模拟步骤。
float time
仿真时间。
float box[3][3]
存储为一组三个基向量的计算盒,以允许三斜PBC。对于矩形框,框的边缘存储在矩阵的对角线上。
3dfcoord x[natoms]
坐标本身以降低的精度存储。请注意,当原子数小于9时,不使用降低精度。
在 C++ 程序中使用XTC
您可以编写自己的分析工具来利用压缩的 .xtc 格式文件:请参阅 安装目录template.cpp中的文件 share/gromacs/template以获取示例,并参阅 https://manual.gromacs.org/current/doxygen/html-full/page_analysistemplate.xhtml 以获取文档。
要读取和写入 xtc 文件,可以通过以下例程获得xtcio.h:
/* All functions return 1 if successful, 0 otherwise */
struct t_fileio* open_xtc(const char* filename, const char* mode);
/* Open a file for xdr I/O */
void close_xtc(struct t_fileio* fio);
/* Close the file for xdr I/O */
int read_first_xtc(struct t_fileio* fio,
int* natoms,
int64_t* step,
real* time,
matrix box,
rvec** x,
real* prec,
gmx_bool* bOK);
/* Open xtc file, read xtc file first time, allocate memory for x */
int read_next_xtc(struct t_fileio* fio, int natoms, int64_t* step, real* time, matrix box, rvec* x, real* prec, gmx_bool* bOK);
/* Read subsequent frames */
int write_xtc(struct t_fileio* fio, int natoms, int64_t step, real time, const rvec* box, const rvec* x, real prec);
/* Write a frame to xtc file */
要使用库函数,请将其包含"gromacs/fileio/xtcio.h" 在您的文件中并链接-lgromacs。
xvg文件
几乎所有 GROMACS 分析工具的输出都可以作为 Grace(以前称为 Xmgr)的输入。我们使用 Grace,因为它非常灵活,而且它还是免费软件。它生成 PostScript™ 输出,非常适合包含在 LaTeX 文档中,也适用于其他文字处理器。
下面显示了使用 GROMACS 数据的 Grace 会话示例:
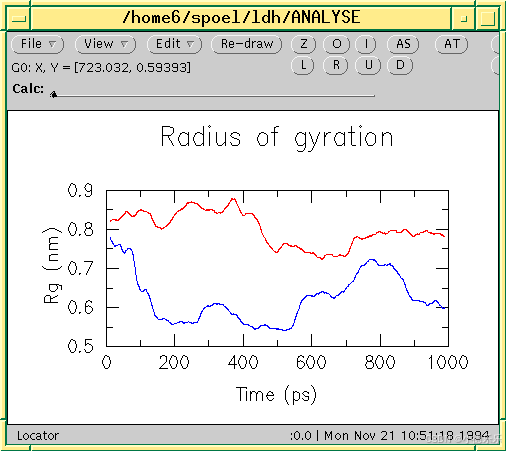
注意
返回主文章:《【分子动力学】 分子动力学新手入门:一文读懂GROMACS使用全流程,轻松开启模拟之旅! GROMACS初学者了解资料 GROMACS安装之前的准备 windows如何安装Gromacs》


















 1258
1258

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









