







1.下载sra文件
wget http……ncbi(data).sra
2.将sra文件转为fastq文件
fastq-dump --split-3 filename
注:- -split-3 如果是双端测序则出现两个fastq文件,如果是单端测序则只有一个文件。得到的文件应该是fastq,下面非为两个方法去找后面的peak。
fasterq-dump --split-3 *.sra -e 12
fasterq-dump可以设置线程 -e
一.nf-core-chipseq pipeline进行分析
说明:该方法适用于有输入文件即input的chipseq分析,如果没有input文件则无法设计design.csv文件
nextflow run /h/jianjin/miniconda3/chipseq-master/main.nf --input /h/jianjin/miniconda3/yy/design.csv --genome GRCh37 --narrow_peak cpus 16
二.正常流程进行chipseq
安装软件:
conda install -y xxx
1.质控
fastqc -t 3 SRR******_1.fastq -o 目录
过程及结果显示
Started analysis of SRR********.fastq
Approx 5% complete for SRR*******.fastq
……
Approx 95% complete for SRR******.fastq
Analysis complete for SRR*******.fastq
2.trim_galore去接头
trim_galore -q 20 --phred33 --stringency 3 --fastqc --length 36 -e 0.1 --paired fq --gzip -o /Data/wu_lab/yuanye/projects/chipseq/daizhongye/2022_6_28_prc2_M/trim/
3.bowtie比对
(bwt) wu_lab@bio:~/yuanye/projects/chipseq/dongfeng/2021_7_27/data/fastq/NPC$ bowtie2 -t -p 10 -x /Data/wu_lab/reference/UCSC_hg19/bowtie2_index/hg19 -U SRR*******_1.fastq.gz -S /Data/wu_lab/yuanye/projects/chipseq/dongfeng/2021_7_27/data/fastq/NPC/result/bowtie2result/SRR******_1.sam
结果显示
Time loading reference: 00:00:00
Time loading forward index: 00:00:01
Time loading mirror index: 00:00:01
Multiseed full-index search: 00:06:31
27524287 reads; of these:
27524287 (100.00%) were unpaired; of these:
1214586 (4.41%) aligned 0 times
17868042 (64.92%) aligned exactly 1 time
8441659 (30.67%) aligned >1 times
95.59% overall alignment rate
Time searching: 00:06:33
Overall time: 00:06:33
注:
(1)使用bowtie的时候我原来在chipseq环境中总是报错,报以下内容的错, 但是当我换了个环境(bwt)这个问题就解决了,具体原因不清楚。(尽量不要在bwt内再安装其他软件了)
但是当我换了个环境(bwt)这个问题就解决了,具体原因不清楚。(尽量不要在bwt内再安装其他软件了)
(2)此次用的是单端测序,因此采用参数-U,如果是双端测序可以看李老师的教程。
(3)双端测序下:
(bwt) wu_lab@bio:~/yuanye/projects/chipseq/dongfeng/2021_8_23/data$ bowtie2 -t -p 16 -x /Data/wu_lab/reference/UCSC_hg19/bowtie2_index/hg19 -1 KD-Hyp-K4me3_FKDL202583604-1a_1.clean.fq.gz -2 KD-Hyp-K4me3_FKDL202583604-1a_1.clean.fq.gz -S /Data/wu_lab/yuanye/projects/chipseq/dongfeng/2021_8_23/bwt/KD-Hyp-K4me3_FKDL202583604.sam
ps:
bowtie2与bowtie1相比,bowtie1适合比对25-50ups的序列read,而bowtie2适合比对100ups左右的序列read,但是Bowtie 2 does not align colorspace reads,而bowtie1可以。
End-to-end alignment versus local alignment
3.sam变bam,bamsort
(chipseq) wu_lab@bio:~/yuanye/projects/chipseq/dongfeng/2021_7_27/data/fastq/NPC/result/bowtie2result$ samtools view -@ 8 -S SRR******_2.sam -b > /Data/wu_lab/yuanye/projects/chipseq/dongfeng/2021_7_27/data/fastq/NPC/result/bam/SRR*****_2.bam
samtools sort -@ 8 -l 5 -o SRR*****.bam.sort SRR****.bam
ps:
使用base环境下,用conda 安装samtools不仅速度慢,而且使用时会出现以下错误。
samtools: error while loading shared libraries: libcrypto.so.1.0.0: cannot open shared object file: No such file or directory
两种解决方法
方法1:
创建一个samtools的独立环境,再用conda 安装
(CHIPSEQ) wu_lab@bio:~/yuanye/projects/chipseq/yuanye/2022_2_16/sam$ conda install samtools openssl=1.0
Collecting package metadata (current_repodata.json): done
Solving environment: done
## Package Plan ##
environment location: /Data/wu_lab/anaconda3/envs/CHIPSEQ
added / updated specs:
- openssl=1.0
- samtools
The following packages will be downloaded:
package | build
---------------------------|-----------------
_openmp_mutex-4.5 | 1_gnu 22 KB conda-forge
curl-7.54.1 | 0 572 KB https://mirrors.tuna.tsinghua.edu.cn/anaconda/pkgs/free
krb5-1.13.2 | 0 3.5 MB https://mirrors.tuna.tsinghua.edu.cn/anaconda/pkgs/free
libdeflate-1.10 | h7f98852_0 77 KB conda-forge
libssh2-1.8.0 | 0 240 KB https://mirrors.tuna.tsinghua.edu.cn/anaconda/pkgs/free
ncurses-5.9 | 10 1.1 MB conda-forge
openssl-1.0.2u | h516909a_0 3.2 MB conda-forge
samtools-1.8 | 4 1.8 MB bioconda
------------------------------------------------------------
Total: 10.4 MB
The following NEW packages will be INSTALLED:
_libgcc_mutex conda-forge/linux-64::_libgcc_mutex-0.1-conda_forge
_openmp_mutex conda-forge/linux-64::_openmp_mutex-4.5-1_gnu
bzip2 conda-forge/linux-64::bzip2-1.0.8-h7f98852_4
ca-certificates conda-forge/linux-64::ca-certificates-2021.10.8-ha878542_0
curl anaconda/pkgs/free/linux-64::curl-7.54.1-0
krb5 anaconda/pkgs/free/linux-64::krb5-1.13.2-0
libdeflate conda-forge/linux-64::libdeflate-1.10-h7f98852_0
libgcc conda-forge/linux-64::libgcc-7.2.0-h69d50b8_2
libgcc-ng conda-forge/linux-64::libgcc-ng-11.2.0-h1d223b6_12
libgomp conda-forge/linux-64::libgomp-11.2.0-h1d223b6_12
libssh2 anaconda/pkgs/free/linux-64::libssh2-1.8.0-0
libstdcxx-ng conda-forge/linux-64::libstdcxx-ng-11.2.0-he4da1e4_12
libzlib conda-forge/linux-64::libzlib-1.2.11-h36c2ea0_1013
ncurses conda-forge/linux-64::ncurses-5.9-10
openssl conda-forge/linux-64::openssl-1.0.2u-h516909a_0
samtools bioconda/linux-64::samtools-1.8-4
xz conda-forge/linux-64::xz-5.2.5-h516909a_1
zlib conda-forge/linux-64::zlib-1.2.11-h36c2ea0_1013
Proceed ([y]/n)? y
Downloading and Extracting Packages
openssl-1.0.2u | 3.2 MB | ######################################################## | 100%
libdeflate-1.10 | 77 KB | ######################################################## | 100%
curl-7.54.1 | 572 KB | ######################################################## | 100%
_openmp_mutex-4.5 | 22 KB | ######################################################## | 100%
libssh2-1.8.0 | 240 KB | ######################################################## | 100%
samtools-1.8 | 1.8 MB | ######################################################## | 100%
krb5-1.13.2 | 3.5 MB | ######################################################## | 100%
ncurses-5.9 | 1.1 MB | ######################################################## | 100%
Preparing transaction: done
Verifying transaction: done
Executing transaction: done
(CHIPSEQ) wu_lab@bio:~/yuanye/projects/chipseq/yuanye/2022_2_16/sam$ samtools -h
[main] unrecognized command '-h'
(CHIPSEQ) wu_lab@bio:~/yuanye/projects/chipseq/yuanye/2022_2_16/sam$ samtools --help
Program: samtools (Tools for alignments in the SAM format)
Version: 1.8 (using htslib 1.8)
Usage: samtools <command> [options]
Commands:
-- Indexing
dict create a sequence dictionary file
faidx index/extract FASTA
index index alignment
-- Editing
calmd recalculate MD/NM tags and '=' bases
fixmate fix mate information
reheader replace BAM header
targetcut cut fosmid regions (for fosmid pool only)
addreplacerg adds or replaces RG tags
markdup mark duplicates
-- File operations
collate shuffle and group alignments by name
cat concatenate BAMs
merge merge sorted alignments
mpileup multi-way pileup
sort sort alignment file
split splits a file by read group
quickcheck quickly check if SAM/BAM/CRAM file appears intact
fastq converts a BAM to a FASTQ
fasta converts a BAM to a FASTA
-- Statistics
bedcov read depth per BED region
depth compute the depth
flagstat simple stats
idxstats BAM index stats
phase phase heterozygotes
stats generate stats (former bamcheck)
-- Viewing
flags explain BAM flags
tview text alignment viewer
view SAM<->BAM<->CRAM conversion
depad convert padded BAM to unpadded BAM
但是该方法的问题就是,如果想一个流程运行下来chipseq还涉及到其他软件在其他环境下的问题,来来回回切换环境很麻烦。
方法二:
创建软连接
(base) wu_lab@bio:~$ cd /Data/wu_lab/anaconda3/lib/
(base) wu_lab@bio:~/anaconda3/lib$ ln -s ~/anaconda3/lib/libcrypto.so.1.1 ~/anaconda3/lib/libcrypto.so.1.0.0
利用命令samtools --help测试软件是否能用。
4.bam.sort.index
(deeptools) wu_lab@bio:~/yuanye/projects/chipseq/dongfeng/2021_8_23/bam$ samtools index -@ 16 Kxxxxxx4.bam.sort
5.查看比对结果
samtools flagstat -@ 8 SRR*****.bam.sort
结果显示
27524287 + 0 in total (QC-passed reads + QC-failed reads)
27524287 + 0 primary
0 + 0 secondary
0 + 0 supplementary
0 + 0 duplicates
0 + 0 primary duplicates
26309701 + 0 mapped (95.59% : N/A)
26309701 + 0 primary mapped (95.59% : N/A)
0 + 0 paired in sequencing
0 + 0 read1
0 + 0 read2
0 + 0 properly paired (N/A : N/A)
0 + 0 with itself and mate mapped
0 + 0 singletons (N/A : N/A)
0 + 0 with mate mapped to a different chr
0 + 0 with mate mapped to a different chr (mapQ>=5)
6.bam转bigwig
(deeptools) wu_lab@bio:~/yuanye/projects/chipseq/dongfeng/2021_8_23/bam$ bamCoverage -p 16 -v -b KD-Hyp-K4me3_FKDL202583604.bam.sort -o KD-Hyp-K4me3_FKDL202583604.bam.sort.bw
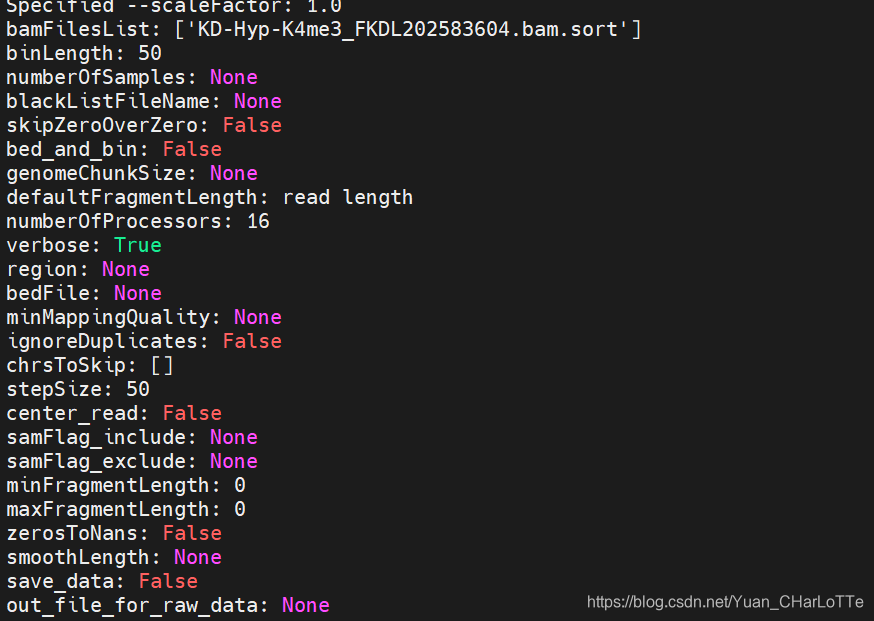
7.MACS peak calling
R-loop数据分析之R-ChIP(peak calling)
(base) wu_lab@bio:~/yuanye/projects/chipseq/dongfeng/2021_7_27/data/fastq/NPC/result$ macs2 callpeak -t /Data/wu_lab/yuanye/projects/chipseq/dongfeng/2021_7_27/data/fastq/NPC/result/bam/SRR*******.bam -f BAM -g hs --outdir /Data/wu_lab/yuanye/projects/chipseq/dongfeng/2021_7_27/data/fastq/NPC/result/macs/ -n SRR****** -B -q 0.01
注:
macs2 callpeak --help
usage: macs2 callpeak -t TFILE # treat组
[-c [CFILE]] # control 或 mock(非特异性抗体,如IgG)组
# control:input DNA,没有经过免疫共沉淀处理;
# mock:1)未使用抗体富集与蛋白结合的DNA片段;2)非特异性抗体,如IgG
[-f] # MACS2读入文件格式,默认自动检测输入文件格式
[-g GSIZE] # 有效基因组大小(可比对基因组大小),人类hs,小鼠mm
[-s TSIZE] # 测序读长;如果不设定,MACS 利用输入的前10个序列自动检测
[–outdir OUTDIR] # MACS2结果文件保存路径
[-n NAME] # 为MACS2输出文件命名
[-B] # 保留the fragment pileup, control lambda, -log10pvalue 和 -log10qvalue scores到bedGraph 文件。
[-q QVALUE | -p PVALUE] # qvalue (minimum FDR)设定call significant regions的阈值;
# 默认,0.01,对于 broad marks(组蛋白修饰的chipseq),可以使用0.05;
# Q-values are calculated from p-values using Benjamini-Hochberg procedure.
# 设定p值时, qvalue不再起作用。
————————————————
版权声明:本文为CSDN博主「垚垚爸爱学习」的原创文章,遵循CC 4.0 BY-SA版权协议,转载请附上原文出处链接及本声明。
原文链接:https://blog.csdn.net/xiaomotong123/article/details/108734627
-t参数指定抗体处理的样本,-c指定input样本,值得一提的是,macs支持多种格式的输入文件,除了上述代码中使用的bam格式外,还支持SAM/BED格式。
–outdir指定输出结果的目录,-n参数指定输出文件名的前缀,-g参数指定基因组的有效大小,在NGS数据中,测序reads在基因组上的覆盖度并不是100%, 而且有些重复区域的比对信息是不可信的,剩下的能够利用的区域通常只占整个基因组区域的70%到90%,这个区域的大小就是有效大小,对于常见的物种,程序内置了有效大小,我们只需要指定物种的缩写即可
————————————————
版权声明:本文为CSDN博主「生信修炼手册」的原创文章,遵循CC 4.0 BY-SA版权协议,转载请附上原文出处链接及本声明。
原文链接:https://blog.csdn.net/weixin_43569478/article/details/108079439
输出文件结果图

如果是broadpeak
macs2 callpeak -t /Data/wu_lab/yuanye/projects/chipseq/dongfeng/2021_8_23/bam/xxxx.bam -f BAM --broad -g hs --outdir /Data/wu_lab/yuanye/projects/chipseq/dongfeng/2021_8_23/peak/ -n xxxx -B --broad-cutoff 0.1
增加-shift 0 --extsize 150(可根据条带适当变大或变小)
-f 输入文件格式 双端加PE 单端加SE或不加
- 双端不建模型(只有单端需要建立模型,具体看这篇MACS2 -m/–mfold使用)
macs2 callpeak --nomodel -t //Data/wu_lab/yuanye/projects/chipseq/daizhongye/2022_6_28_prc2_M/H3K27me3_EZH2/bam/${i}_rmdup.bam.sort -f BAMPE -g mm --outdir /Data/wu_lab/yuanye/projects/chipseq/daizhongye/2022_6_28_prc2_M/H3K27me3_EZH2/peak/p_0.01/ -n ${i} -B -p 0.01
peak结果文件解读
narrow,broad, gapped peak:三种格式之间的区别与联系
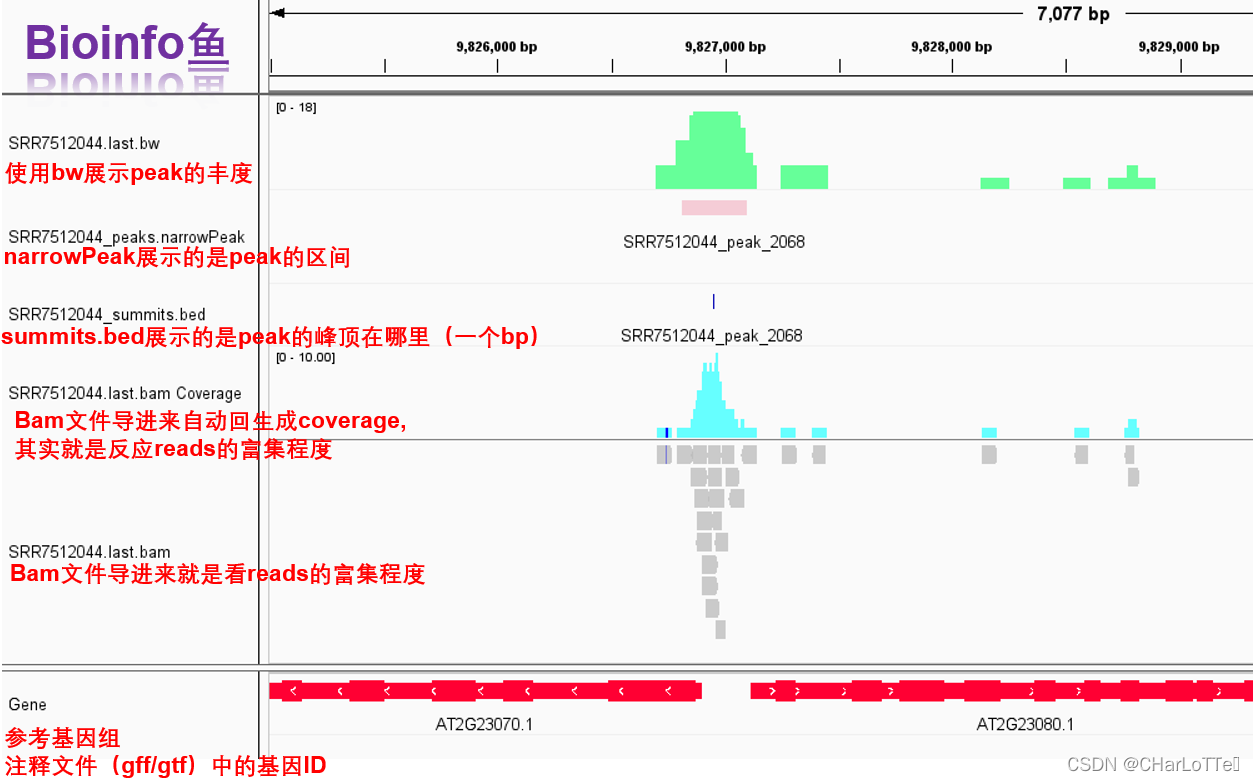
8.可视化
https://www.jianshu.com/p/9aa719faa4b5

















 1471
1471

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









