







1.QC质控 步骤与CHIPSEQ相同
2.STAR进行比对
安装
wget https://github.com/alexdobin/STAR/releases/tag/STAR_2.4.2a
tar -xzf STAR_2.4.2a.tar.gz
cd STAR_2.4.2a
# Build STAR
cd source
make STAR
这里是引用
2.1建立STAR索引
建立索引前首先下载参考基因组文件
人类参考基因组网站
https://www.gencodegenes.org/human/
 ↑下载GTF文件
↑下载GTF文件
 以及fa文件。
以及fa文件。
利用wget -c可以无断点下载。
利用wget -i 下载.txt文件
鼠的基因组同上。
- hg19的基因组下载时下载fasta文件时,下下面两个文件

 并将transcript文件放到starindex文件夹下,为后面rsem建索引做准备。
并将transcript文件放到starindex文件夹下,为后面rsem建索引做准备。
建立索引时
STAR --runMode genomeGenerate --runThreadN 16 --genomeDir /Data/wu_lab/reference/starindex/mouse/ --genomeFastaFiles /Data/wu_lab/reference/starindex/mouse/GRCm39.genome.fa --sjdbGTFfile /Data/wu_lab/reference/starindex/mouse/gencode.vM27.annotation.gtf
显示successfully即为成功
2.2使用STAR
STAR --runThreadN 16 --runMode alignReads --readFilesCommand zcat --quantMode TranscriptomeSAM GeneCounts --outSAMunmapped None --genomeDir /Data/wu_lab/reference/starindex/mouse/ --readFilesIn /Data/wu_lab/yuanye/projects/RNAseq/daizhongye/2021_8_12/data/xxxxxx.fq.gz --outFileNamePrefix xxxxxx
比对过程:
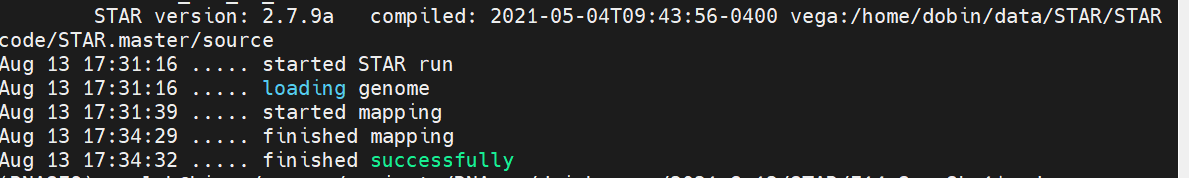 比对结果文件:
比对结果文件:

双端比对时 输入文件该文${i}_1.fq.gz和 ${i}_2.fq.gz
其中
EZH2-RA3h-rep2Log.final.out
文件里保存的是比对率等信息,可以查看比对统计信息。
以"Aligned.sortedByCoord.out.bam"为后缀的BAM文件中,reads比对到的位置是基因组位置。
以"Aligned.toTranscriptome.out.bam"为后缀的BAM文件中,reads比对到的位置是转录本位置。
https://www.bioinfo-scrounger.com/archives/288/
3.rsem转录本定量
安装
conda install -c bioconda rsem
3.1 建索引
(RNASEQ) wu_lab@bio:~/reference/starindex/mouse$ rsem-prepare-reference --gtf mm9.gtf --STAR GRCm39.genome.fa -p 30 /Data/wu_lab/reference/mousersemindex/mouse/
建立过程及结果显示
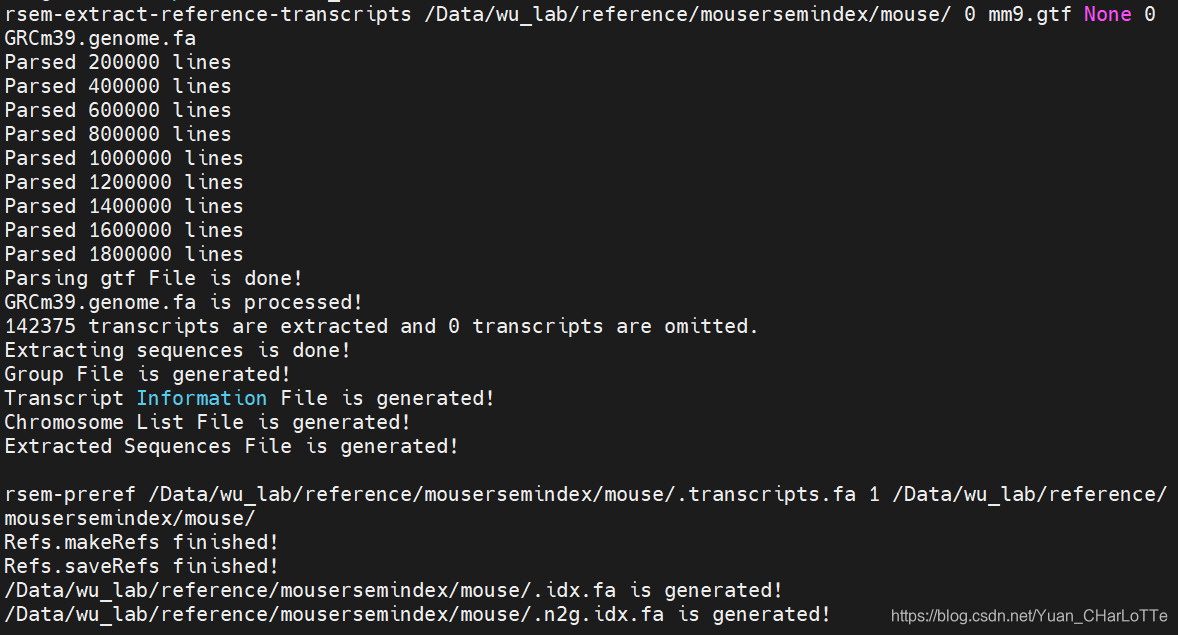
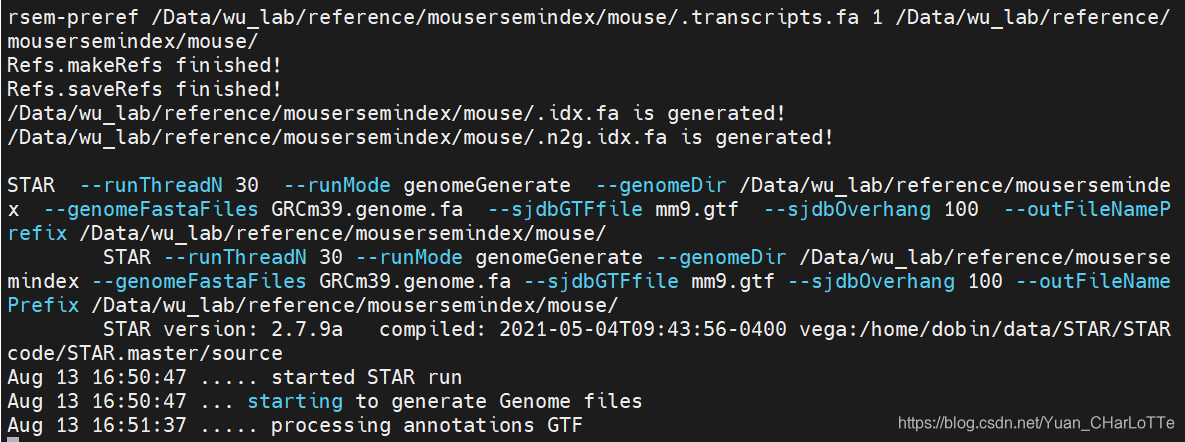
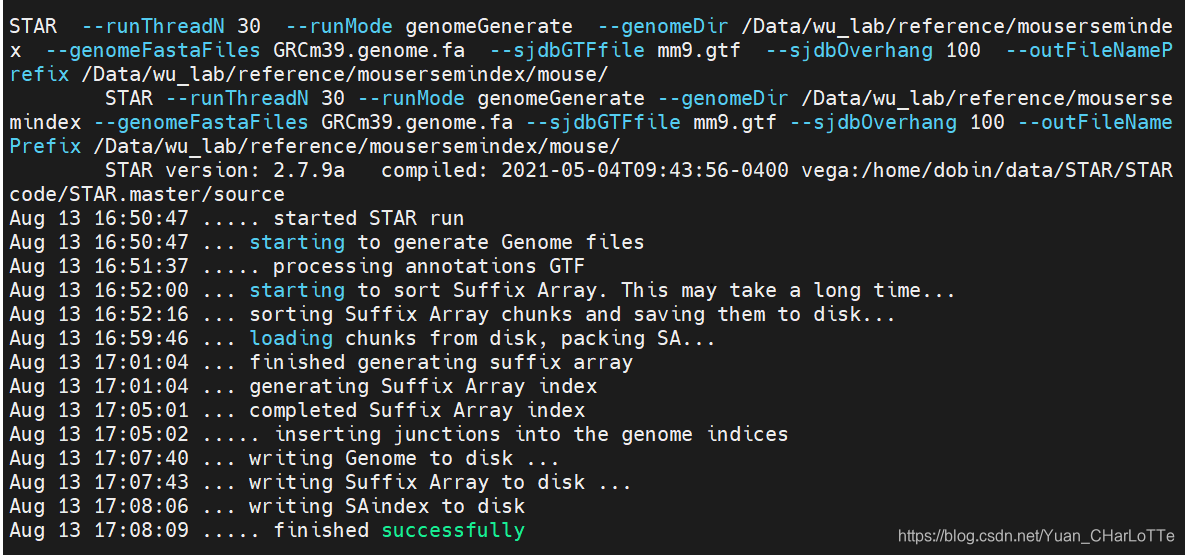
3.2 运行rsem
(RNASEQ) wu_lab@bio:~/yuanye/projects/RNAseq/daizhongye/2021_8_12/rsem/E14_Scr_SL_1$ rsem-calculate-expression -no-bam-output --alignments -p 30 /Data/wu_lab/yuanye/projects/RNAseq/daizhongye/2021_8_12/STAR/xxxxxx/xxxxxAligned.toTranscriptome.out.bam /Data/wu_lab/reference/mousersemindex/mouse/ xxxx
运行过程
双端比对时
rsem-calculate-expression --paired-end -no-bam-output --alignments -p 8 input_Aligned.toTranscriptome.out.bam reference_name out_prefix
















 7026
7026











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









