







 本文详细介绍了ChIP-seq数据分析的全流程,包括SRA文件下载、adaptor去除、质量控制、参考基因组比对、BAM文件统计、PCR去重、MACS2找peak、Deeptools可视化及ChIPSeeker注释。通过这个教程,读者可以掌握ChIP-seq数据的基本分析方法。
本文详细介绍了ChIP-seq数据分析的全流程,包括SRA文件下载、adaptor去除、质量控制、参考基因组比对、BAM文件统计、PCR去重、MACS2找peak、Deeptools可视化及ChIPSeeker注释。通过这个教程,读者可以掌握ChIP-seq数据的基本分析方法。
前言
不知不觉,时间过得太快,这个教程的学习居然拖。。。了半年!
趁春节放假,还是把它完成吧。顺完这个教程之后,测序相关的上下游分析算是打通了一二,暂告一段落。
参考Jimmy老师的ChIP-seq教程《https://cloud.tencent.com/developer/article/1346037》。感恩!
过程不赘述,上面教程有所有的分析和代码。
还是在conda里面进行分析,这个环境集中安装了所有需要的程序。

简略过程
如果你做过RNA-seq或者WES等的上游分析,那流程应该就很熟悉了。
1 下载SRA文件
#开代理吧,网速飞起
fasterq-dump --split-3 -O ../fastq $id/$id.sra
2 去掉adaptor
trim_galore $fq1 -q 25 --phred33 --length 25 -e 0.1 --stringency 4 -o ../clean
3 qc质量控制
## qc reports
cd ../qc
ls ../raw/*fastq|xargs fastqc -t 10 -o ./
multiqc ./
ls ../clean/*fq|xargs fastqc -t 10 -o ./
multiqc ./
4 与参考基因组比对
file=$(basename $id)
sample=${file%%.*}
bowtie2_index=~/public/references/index/bowtie2/mm10/mm10
bowtie2 -p 5 -x $bowtie2_index -U $id | samtools sort -O bam -@ 5 -o - > ${sample}.bam
5 查看Bam文件统计信息
ls *.bam |xargs -i samtools index {}
ls *.bam | while read id ;do ( samtools flagstat $id > $(basename $id ".bam").stat & );done
6 合并Bam
ls *.bam|sed 's/_[0-9].bam//g' |sort -u |while read id;do samtools merge -f ../mergeBam/$id.merge.bam $id*.bam ;
7 PCR去重
ls *merge.bam | while read id ;do (nohup samtools markdup -r $id $(basename $id ".bam").rmdup.bam & );done
8 使用macs2 找 peak
nohup macs2 callpeak -c Control.merge.bam -t $id.merge.bam -f BAM -B -g mm -n $id --outdir ../peaks 2> $id.log &
这里找peak,是需要和control组进行比较的,也就是找差异。
得到下面的*_summits.bed就是与control组比较完之后的差异序列信息。
可以看到control组为零。


找完peak,上游分析就结束了。下面进行一些可视化的探索。
9 使用Deeptools进行
## change to bw file
refgene=~/public/references/CHIPSeq/mouse.ref
samtools index $id # create indexs
bamCoverage -b $id -o $sample.bw
computeMatrix reference-point --referencePoint TSS -p 4 -b 2000 -a 2000 -R $refgene -S $sample.bw --skipZeros -o ${sample}_TSS.gz --outFileSortedRegions ${sample}_genes.bed
plotHeatmap -m ${sample}_TSS.gz -out ../plotHeatmap/${sample}.pdf --plotFileFormat pdf --dpi 720
10 单独和所有组别画图

可以看到单独画图和一起画图的区别。这种图大概属于CNS系列,高大上。
11 R里面进行ChIPSeeker注释
library(ChIPseeker)
bedPeaksFile = 'H3K36me3_summits.bed';
#bedPeaksFile
require(ChIPseeker)
require(TxDb.Mmusculus.UCSC.mm10.knownGene)
txdb <- TxDb.Mmusculus.UCSC.mm10.knownGene
require(clusterProfiler)
peak <- readPeakFile( bedPeaksFile )
keepChr= !grepl('_',seqlevels(peak))
seqlevels(peak, pruning.mode="coarse") <- seqlevels(peak)[keepChr]
peakAnno <- annotatePeak(peak, tssRegion=c(-3000, 3000),
TxDb=txdb, annoDb="org.Mm.eg.db")
peakAnno_df <- as.data.frame(peakAnno)
得到下面的注释:
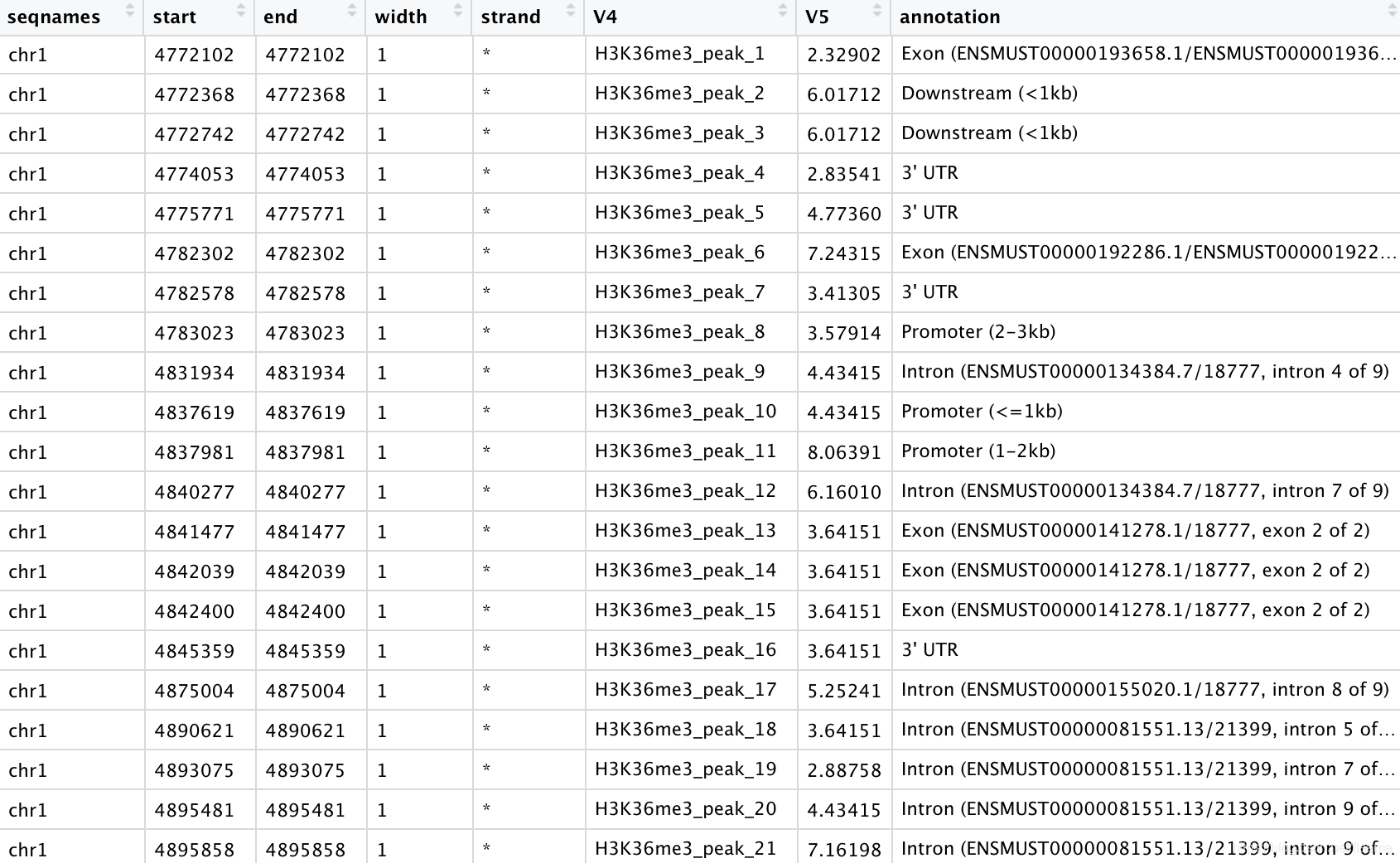
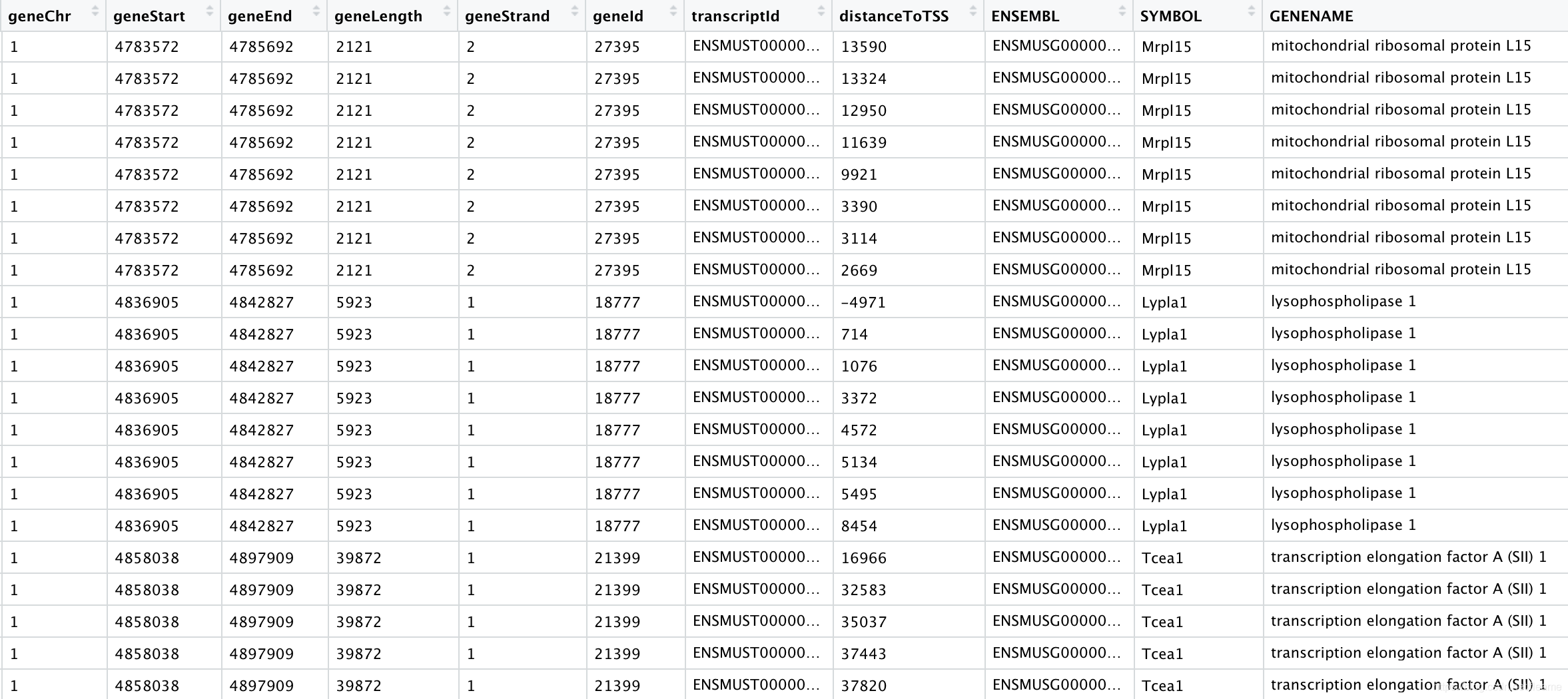
其他的下游分析可自行尝试。
好了,这个教程学习到此结束。下一个目标,单细胞测序分析!我这步子虽然迈了慢一点,但我坚持下来了。
文章首发于微信公众号:颗粒神经元。欢迎关注。

















 8221
8221

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









