






语言模型在人工智能(AI)和计算生物学的许多领域正发挥着日益重要的作用。在本入门指南中,我们讨论了基于自然语言和基于生物序列的语言模型如何应用于生物研究。本文主要面向希望在自身研究中应用这些前沿AI技术的生物学家,提供适应生物学需求的语言模型最佳实践指南和关键资源。
语言模型是一种能够学习序列中复杂模式的人工智能方法,例如句子中的单词或蛋白质中的氨基酸。近年来,由于ChatGPT等文本生成式大模型的推动,这类模型获得了广泛关注。这些模型通过在海量异质数据集合的训练,学习到了很多模式,并可被调整以解决各种特定问题。例如,ChatGPT最初被训练用于补全文本中的缺失词汇,但该训练过程使其具备了语言推理能力,能够处理从总结论文到编写生物信息学代码等多样化任务。
此外,语言模型能够应用于解决其初始设计目标之外的问题,甚至优于专门针对这些问题训练的模型。得益于这种灵活性,语言模型通常作为基础模型,支撑广泛的下游应用。这类模型不仅限于自然语言(如英语),还可处理由生物实体(如氨基酸或基因)序列构成的生物语言。
本文讨论的语言模型均基于Transformer架构——一种能够捕捉长序列模式的人工神经网络(关键术语表见框1)。例如,它可以学习论文摘要如何总结后续文本,或识别蛋白质序列中相距较远的氨基酸之间的物理互作。这类模型通常采用无监督的生成式训练,即训练序列不带有特定标签(无监督),而是通过重构输入序列进行学习(生成式)。
通过这种生成过程,模型学习数据形式背后的潜在规则。为了让模型实现其他目标,通常会在已预训练模型的基础上,使用新数据进一步训练(微调)。这一过程称为迁移学习——通过大规模数据集预训练赋予模型对数据的基础理解,使其在微调阶段能更高效地学习新任务。
术语表
术语 | 定义 |
语言模型 | 基于训练数据学习、预测序列中其他元素出现概率的AI模型。 |
自然语言模型 | 基于人类语言文本序列训练的语言模型。 |
生物语言模型 | 将氨基酸、基因等生物实体视为”语言”训练的语言模型 |
基础模型 | 预训练获得的、可灵活适配多种应用的AI模型 |
Transformer | 通过注意力机制动态权衡输入数据不同部分重要性的序列处理模型 |
标记 | 序列的基本单元(如单词、词根、氨基酸、基因),作为模型的输入单位 |
嵌入表示 | 模型学习到的输入数据的数值表示(一个数字列表或数字向量),捕捉输入文本的语义或功能特征 |
迁移学习 | 将预训练模型调整用于相关任务的机器学习方法,通常需要更少数据和训练时间 |
预训练 | 迁移学习前的第一步,通过大规模数据训练模型学习通用特征 |
微调 | 迁移学习的第二步,通过较小规模的特定任务数据调整或对接预训练模型 |
多模态模型 | 可处理并整合多种输入数据类型(如文本与图像)的模型。 |
生成式模型 | 被训练用于生成与训练数据类似的新数据的模型。 |
无监督学习 | 一种从无明确标签的数据集中学习的机器学习方法,常用于发现数据中的模式或结构。 |
幻觉生成 | 语言模型生成看似合理但事实错误或前后矛盾的输出。 |
降维 | 在保留数据核心结构的前提下减少高维数据特征数量的技术,常用于将模型嵌入降至二维以进行可视化。 |
掩码 | 用于替换输入序列某部分的特殊词元,模型基于上下文学习预测其内容。 |
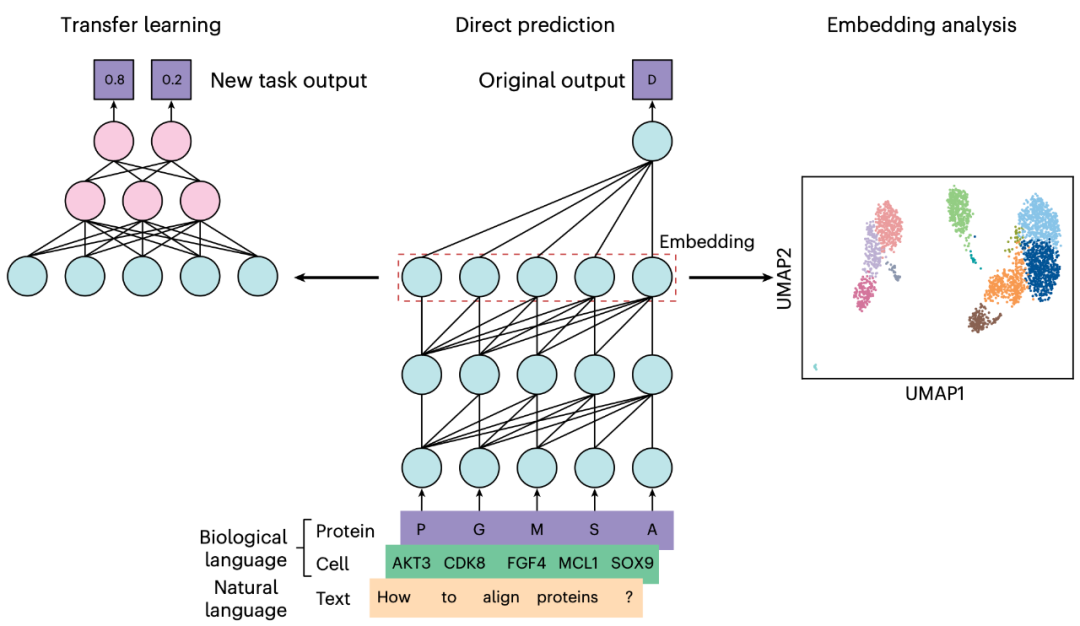

图1 | 语言模型在生物研究中的应用方法。用于生物研究的语言模型可基于自然语言(如英语)或生物语言(如蛋白质序列或单细胞数据的一串基因)。无论哪种情况,输入数据均被分解为称为“词元”的单元(如单词或氨基酸),并由模型处理。随后可通过以下三种方法之一将语言模型适配于生物研究。在迁移学习方法(左)中,预训练模型可选择性调整(如扩展模型结构),并通过进一步训练(微调)解决其初始未涵盖的特定任务。在直接预测方法(中)中,模型直接基于输入数据进行预测,例如预测句子中的下一个单词。在嵌入(embedding)分析方法(右)中,模型计算的输入表征(即嵌入表示)被用于降维或聚类等分析任务。
在本入门指南中,我们将探讨如何通过上述三种方法调整自然语言模型与生物语言模型,以解决生物学中的各类问题。同时,我们亦讨论生物学领域使用语言模型的最佳实践,包括不同方法的适用场景建议,以及需结合当前模型局限性考虑的注意事项。
自然语言模型
生物学自然语言模型简介
尽管生物学本质上依赖于蛋白质、基因和细胞等物理实体的特性,但我们对这一领域的理解是通过科学论文、教科书、网页等自然语言的形式记录的。因此,利用自然语言模型帮助生物学研究者便捷获取这些文本资源中蕴含的海量生物信息,正受到越来越多的关注。此外,自然语言模型可通过整合图像或基因序列等多模态数据进行增强,形成多模态模型,从而深入解析不同形式的生物实体。下文我们将聚焦直接预测方法,探讨自然语言模型在生物研究中的应用。
生物学通用模型与专业模型对比
自然语言模型可分为通用模型(如ChatGPT或Claude),其训练数据涵盖包括PubMed等生物医学资源在内的广泛文本语料库。也可设计为专业模型(如BioBERT或Med-PaLM),这类模型专门针对生物医学文本进行训练或微调。当前通用模型凭借其广泛的知识储备,在回答医学考试问题等生物医学任务中表现优于专业模型。
自然语言模型在生物学文献理解中的应用
自然语言模型的核心优势在于其能够基于海量生物学文献进行推理,并将信息提炼为易于理解的回答。例如,若研究者遇到不熟悉的技术概念(如多序列比对),可直接要求语言模型用简洁的语句解释该概念,无需自行查阅资料。研究者还可根据自身背景调整输入(如“向具有生物学入门背景的人解释多序列比对”),以获取量身定制的答案。除总结生物学概念外,自然语言模型还可帮助研究者快速理解新科学内容。例如,科学家可向模型提供新论文链接,要求其总结内容或回答关于论文方法的具体技术问题(图2)。此类模型甚至能基于现有文献提出新的生物医学研究思路(例如利用AI处理多序列比对的新方法)。
Input: “Explain the role of CRISPR in genome editing.”
Output: “CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) is a bacterial immune system adapted for precise genome editing. The CRISPR-associated protein Cas9 acts as molecular scissors…”
尽管自然语言模型是理解生物学文献的强有力工具,但其存在一个显著局限——易产生“幻觉”,即生成看似连贯但包含事实错误的文本。此外,这些模型可能不会批判性评估其处理的内容,可能直接反映作者的解读而未质疑其是否得到数据支持。因此,必须对模型输出的准确性进行事实核查,并批判性评估其得出的任何结论。
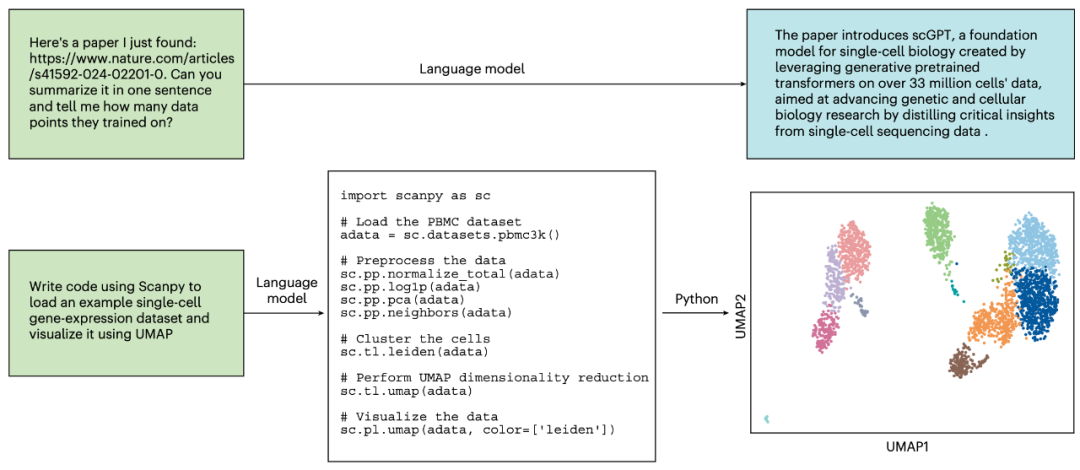
图2 | 自然语言模型在生物研究中的使用示例。在与语言模型的示例对话中,用户向模型提问(绿色文本框),模型则以回答(蓝色文本框或白色代码片段)响应。顶部示例中,用户要求获取科学论文的相关信息,语言模型通过查找、阅读并总结论文内容给出回答。底部示例中,用户请求生物信息学分析的示例代码,语言模型提供可运行的Python代码以生成数据可视化分析。顶部示例由ChatGPT的GPT-4模型生成,底部示例由Claude 3生成(为简洁性稍作编辑)。
自然语言模型在软件交互中的应用
除理解科学文献外,自然语言模型还可通过协助科学家与软件交互(包括编写与调试代码)加速研究进程。此类模型蕴含丰富的生物信息学分析知识,可帮助研究者编写数据处理、结果分析、绘图等任务的代码。当研究者不熟悉领域特定软件包(如用于单细胞分析的Scanpy)时,这些模型尤为实用——它们既了解何时应使用这些工具包,也掌握如何与各组件交互(图2)。自然语言模型还可作为高效的调试工具:向其提供错误代码或报错信息,即可要求其生成修正后的代码。此外,这些模型可为需领域专业知识操作的软件工具提供自然语言接口。例如,ChemCrow允许用户用自然语言提出问题(如“设计一种可溶性分子”),并通过调整用户查询使ChatGPT能够调用化学专用软件工具(如分子合成路径规划)。这种能力使更广泛的科学用户群体能够便捷使用这些工具。
生物语言模型
自然语言模型被训练用于生成文本,而生物语言模型则被训练用于生成生物数据(如氨基酸序列)。本文将探讨采用此方法的两个典型生物语言模型:蛋白质语言模型与单细胞语言模型。尽管聚焦于这两类示例,但生物语言模型的应用范畴并不受限,任何可表示为序列形式且拥有大规模数据集的生物实体(如DNA)均可适用。
蛋白质语言模型
- 为何需要大规模预训练?
基于海量蛋白质序列数据集预训练的蛋白质语言模型,能够学习捕捉进化约束与蛋白质关键特性的表征。通过对这些模型在较小标注数据集上进行后续微调,可准确预测蛋白稳定性、蛋白相互作用等下游任务,甚至设计具有特定结构的蛋白序列。
蛋白质语言模型示例:ESM-2。 ESM-2即为此类模型之一。该模型为Transformer神经网络,基于超过2.5亿条蛋白质序列,通过预测随机遮蔽的氨基酸进行训练。在训练过程中,序列中一部分氨基酸被随机替换为虚构的“掩码”氨基酸,模型需预测被遮蔽的原始氨基酸。通过准确预测特定序列上下文中应出现的氨基酸,模型学习到支配蛋白质结构与功能的模式和约束。正如存在多种自然语言模型,蛋白质语言模型也有多种训练范式。例如,部分模型可能针对特定蛋白质家族的数据进行训练,或采用类似自然语言模型的顺序预测方式预测氨基酸,而非随机遮蔽。
应用:直接预测。根据其原始训练目标,这些模型可直接预测特定序列位置出现每种氨基酸的概率。由于训练数据涵盖所有已知功能性蛋白质序列,模型能有效学习蛋白质进化模式。即使缺乏突变影响的实验数据,模型仍可隐式推断哪些突变会损害蛋白质功能——因为根据进化过程中经验观察的突变规律,这些突变被认为极不可能发生。因此,此类预测可直接用于评估蛋白质编码突变的影响。
具体操作中,可通过遮蔽特定位置的野生型氨基酸,要求模型基于剩余序列推测被遮蔽位点可能是哪些氨基酸,从而获得突变氨基酸的可能性。若模型预测其它突变氨基酸的可能性低于野生型氨基酸,则表明该突变可能有害。基于模型预测可能性的实验研究已验证此类评估方法与突变致病性的相关性。
将蛋白质序列建模为语言的一大优势在于,这些序列无需先进行比对或注释;而其他方法需依赖进化比对后的蛋白质序列才能预测突变影响。语言模型对蛋白质序列可能性的预测还可用于评估序列是否可能形成功能性结构,这使得蛋白质语言模型能够评估和设计新序列。
应用:嵌入分析。 除输出结果外,蛋白质语言模型还可生成蛋白质的嵌入表示。具体而言,当蛋白质序列输入模型处理后,可提取模型对序列中每个氨基酸的内部表征(嵌入)。这些氨基酸嵌入既可单独使用,也可整合为单一蛋白质表征。例如,先前研究发现,对蛋白质序列嵌入进行聚类可识别同源蛋白质,进而基于各蛋白质中氨基酸嵌入的相似性构建多序列比对。
应用:迁移学习。 模型学习的表征可用于解决更具体的任务。由于微调蛋白质语言模型成本较高,许多应用将模型生成的嵌入作为输入,训练更小规模的下游任务模型。例如,此类嵌入已被用于预测蛋白质稳定性、病毒抗原突变的免疫逃逸,以及基于少量标注数据评估错义变异的致病性。此外,新兴高效微调技术使得计算资源有限的研究者也能对大型蛋白质语言模型进行完整微调。
交互式示例。 为展示蛋白质语言模型如何应用于下游任务,我们提供了一个交互式笔记本(使用ESM-2模型),包含直接预测、嵌入分析与迁移学习的实例。用户可通过Google Colab在浏览器中运行:
https://colab.research.google.com/drive/1zIIRGeqpXvKyz1oynHsyLYRxHHiIbrV5?usp=sharing.蛋白质结构模型。 尽管蛋白质结构预测模型(如AlphaFold2和ESMFold2)并非本指南重点,但值得指出的是:像结构预测模型那样,将结构信息与蛋白质序列结合用于训练,可提升下游任务中蛋白质表征的质量。与语言模型类似,蛋白质结构预测模型已证明能通过直接预测、嵌入分析和迁移学习广泛适配多样化下游应用。
单细胞语言模型
为何需要大规模预训练? 单细胞基因表达数据可揭示单个细胞的生理状态与功能,但其高维特性也使对其解读充满挑战。近期开发的AI方法助力分析此类复杂数据。随着公开可用的单细胞基因表达数据激增,研究者得以在数百万跨细胞类型、组织、生物体和实验条件的转录组数据上训练语言模型。这些模型能泛化至新数据集,并可通过微调适应细胞类型注释、批次效应校正等下游任务。
单细胞语言模型示例:Geneformer。 Geneformer即为此类模型之一。与许多其他生物语言模型类似,其采用Transformer架构训练,旨在为下游应用提供基因与细胞的表征。Geneformer将每个细胞表示为该细胞中表达量最高的
2,048个基因的列表,并按RNA表达丰度排序。其训练过程与前述蛋白质语言模型相似:随机遮蔽部分基因,模型通过预测被遮蔽基因进行训练。为了根据表达水平顺序准确预测缺失基因,模型需理解不同基因表达水平的相互作用,并隐式学习细胞类型特异性模式与上下文关系。Geneformer基于涵盖40种组织类型的3,000万个单细胞转录组数据进行训练,从而学习多样化的表达模式。尽管Geneformer关注基因的相对表达水平,其他单细胞语言模型采用不同范式。例如,单细胞语言模型scGPT通过预训练定量表达值实现差异化的下游应用,并可整合实验模态、批次及扰动条件等元数据。应用:直接预测。单细胞语言模型的直接输出支持多种创新的计算机模拟实验。模型可通过调整按表达量排序的细胞基因列表顺序,量化输出变化,从而估计基因扰动对细胞的影响。例如Geneformer通过人为将POU5F1、SOX2、KLF4和MYC基因提升至细胞基因排序顶端,模拟了成纤维细胞重编程过程,从而通过计算将细胞推向诱导多能干细胞状态。类似地,单细胞语言模型还可通过人为删除细胞排序列表中的基因并观察对细胞嵌入的影响,预测细胞对基因移除的敏感性。
应用:嵌入分析。 单细胞语言模型生成的每个基因嵌入可被整合(如取平均值)为单个细胞的整体表征。此类细胞嵌入可用于聚类、可视化及细胞类型标记。得益于训练数据的多样性与规模,这些模型能在保留生物学变异的同时隐式减少批次效应,从而从包含多批次实验的数据集中识别精细的细胞亚型。
应用:迁移学习。 尽管这些嵌入中可自然涌现出有意义的分簇(如细胞类型),但模型仍可通过微调预测单个细胞的特性。例如,单细胞语言模型可微调以整合跨实验条件的数据,预测细胞类型标签与细胞状态。此类模型甚至支持基因的多模态表征。例如,scGPT可通过微调将染色质可及性、蛋白质丰度与基因表达水平结合,实现跨模态数据集的整合。
生物学多模态语言模型
多模态模型能够跨多种数据模态(如文本与图像)进行推理,从而赋能模型解决本质上涉及多类型数据的任务。例如,病理-语言图像预训练模型(PLIP)基于推特数据训练,可将病理图像与其描述文字匹配,用户既可通过图像获取描述,也能通过文字检索对应图像。类似地,Med-PaLM Multimodal被训练用于基于生物医学图像回答问题;MolT5则根据分子结构,以自然语言描述分子特性(包括潜在生物学功能)。若有充分的多模态数据样本,研究者也可针对其他类型的生物数据训练多模态模型。
自然语言模型也可通过结合固定文本嵌入与其他领域数据,实现无需额外训练的多模态应用。 GenePT即以此方式应用于单细胞数据。该模型利用语言模型的隐含基因组学知识生成细胞嵌入:首先通过ChatGPT对NCBI基因文本描述进行嵌入,再按单细胞表达水平加权平均,生成单细胞嵌入表示。某些应用中,此类自然语言模型生成的嵌入效果优于或持平于生物语言模型(如Geneformer)的嵌入。类似思路可推广至生物学其他领域:固定语言模型嵌入可无需额外训练,直接与其他模态的数据或模型整合。
生物学语言模型使用最佳实践
自然语言模型与生物语言模型在生物学研究中具有广泛用途。我们已探讨了三种将其应用于下游研究问题的方法:直接预测、嵌入分析与迁移学习。本文将概述如何根据具体研究问题选择合适方法。最佳方法取决于研究目标、可用数据及计算资源。图3总结了语言模型使用的核心决策流程。尽管这些规则并非绝对,但可为实践提供指导。
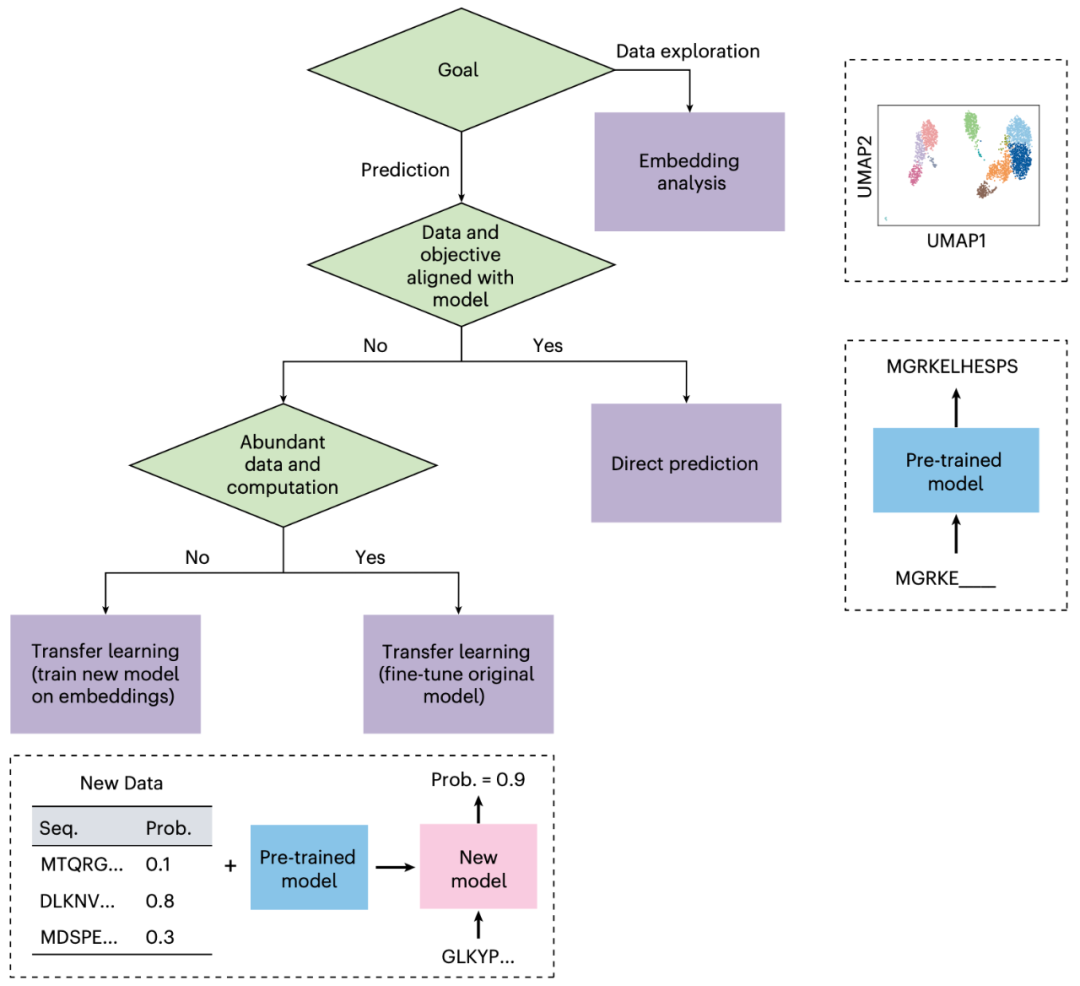
图3 | 选择适配语言模型的合适方法。 确定语言模型最佳使用方式的核心决策流程。语言模型可通过直接预测、嵌入分析或迁移学习适配于生物研究。
对于数据探索,嵌入分析方法可基于模型生成的嵌入对数据进行可视化。
对于预测任务,方法的选择取决于新问题的数据与目标。
若新问题的数据和目标与原始训练目标一致,则适用直接预测(可能需要调整模型输入以优化预测结果)。
若新问题与原始目标不完全匹配,迁移学习更为有效——包括对原模型(通常较大)进行微调,或利用语言模型嵌入作为输入训练新模型(通常较小)。
微调原模型通常效果更强,但相比基于嵌入训练新模型,需更多数据与计算资源。
首要决策是明确研究问题的目标。
若目标为数据探索,可通过嵌入分析方法结合降维、聚类等技术揭示数据结构。
若目标是从数据中做出预测,则直接预测与迁移学习方法更为适用。
当数据与计算资源充足时,最佳策略可能是对语言模型进行部分或全部微调。
若资源有限,则可计算新数据的嵌入表示,并以此作为输入训练一个独立的小型模型。
若研究问题与模型的训练数据及目标内在能力匹配,则直接预测方法适用(可能需要根据目标调整输入)。
若项目目标与模型能力存在显著差异,或存在更贴合任务的特定数据,迁移学习更为有效。
此外,部分模型仅以网页接口或API形式提供,可能限制其用途为直接预测。而拥有开源代码及预训练参数的模型,则支持嵌入分析或迁移学习应用。
语言模型可通过多种方式访问与调整。例如,部分模型提供用户友好的网页界面直接进行预测;其他情况下,代码与训练模型可从Hugging Face或GitHub下载。某些模型附有Jupyter或Google Colab交互式笔记本,展示如何将预训练模型应用于不同场景。若无此类笔记本,包含模型代码的GitHub仓库通常提供文档或示例代码。表1列出的部分应用已配备此类网站与笔记本资源。
表1 | 生物语言模型资源
名称 | 简介 | 资源位置 | 使用方法 |
|---|---|---|---|
ChatGPT | 自然语言(通用型) | https://chat.openai.com | 通过chat.openai.com直接对话,或按文档说明通过API编程调用。 |
DeepSeek | 自然语言(通用型) | https://www.deepseek.com | 通过https://www.deepseek.com直接对话,或按文档说明通过API编程调用。 |
BioBERT | 自然语言(生物医学专用) | GitHub链接 | Hugging Face仓库提供不同规模的预训练BioBERT模型及微调代码。 |
Med-PaLM 2 | 自然语言(生物医学专用) | Google Vertex AI平台 | 作为Google Vertex AI平台组件,向特定客户开放。 |
ESM | 蛋白质语言模型 | GitHub链接 | 提供模型代码、预训练模型及教程资源。 |
ProGen | 蛋白质语言模型 | GitHub链接 | 提供预训练模型及预测代码。 |
Geneformer | 单细胞语言模型 | Hugging Face链接 | 通过Hugging Face获取训练好的模型,文档含多种应用示例代码。 |
scGPT | 单细胞语言模型 | GitHub链接 | 提供预训练模型及包含细胞注释等功能的网页工具。 |
GenePT | 多模态(自然语言+单细胞基因表达) | GitHub链接 | 提供预计算基因嵌入及多种应用的交互式教程。 |
PLIP | 多模态(自然语言+病理图像) | Hugging Face链接 | 包含训练数据、预训练模型及代码的完整资源。 |
Hugging Face | 多类型语言模型平台 | Hugging Face平台 | 搜索特定领域关键词(如“protein”)获取相关模型,支持多种机器学习框架。 |
尽管语言模型功能强大,但用户需注意其重要局限性。
首先,语言模型仍无法完美解决许多生物学问题,甚至包括其原始训练目标相关的任务。原因可能在于
自然语言模型仅涵盖训练数据中的生物学知识,因此无法获知训练后新发现的成果;
蛋白质语言模型通常基于标准氨基酸训练,其输入表征无法体现翻译后修饰的重要性;
单细胞表达数据可能含噪声,且资助导向可能导致数据集中于特定组织类型或疾病状态,这些因素均可能影响模型性能。
模型本身的局限性(无法完全学习训练数据中的所有规律)或训练数据的缺陷。
训练数据可能过时、存在噪声,或缺乏某些类型数据的充分覆盖。例如,
此外,针对特定生物应用设计的模型有时仍优于生物语言模型——尤其是在模型设计可融入先验知识时。例如,整合蛋白质结构信息的方法已被证明优于仅基于蛋白质序列训练的语言模型。最后,评估语言模型在适配其他任务时的性能也需谨慎。语言模型基于未公开的海量数据训练,因此难以确保其训练数据与下游任务测试数据间无信息泄露 (指代测试数据已被应用于模型训练的情况)风险。
结论
本入门指南阐述了如何利用语言模型(包括自然语言模型如ChatGPT,以及生物语言模型如ESM-2和Geneformer)推动生物研究。这些模型通过直接预测、嵌入分析和迁移学习支持广泛的生物学应用。尽管语言模型可辅助生成假设并帮助解释结果,但其仍会出错且尚无法替代实验验证。语言模型还需大量数据训练,目前其生物学应用限于已有大规模数据集的领域。随着更多数据被生成和公开共享,我们预计语言模型将影响生物学中更多样化的领域。
代码可用性
蛋白质语言模型的交互式示例代码可通过Google Colab运行,访问链接:
https://colab.research.google.com/drive/1zIIRGeqpXvKyz1oynHsyLYRxHHiIbrV5?usp=sharing
相关代码及数据亦发布于GitHub:
https://github.com/swansonk14/language_models_biology
高颜值免费 SCI 在线绘图(点击图片直达)
最全植物基因组数据库IMP (点击图片直达)

往期精品(点击图片直达文字对应教程)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
















































 414
414

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











