







为什么没有(四)?
(四)主要说的就是SAM格式,网上一搜就有,就没必要了
(五)就草草地记录了Chapter 4.1: RNA-Seq Applications - Chapter 5.2 Differential RNA-Seq
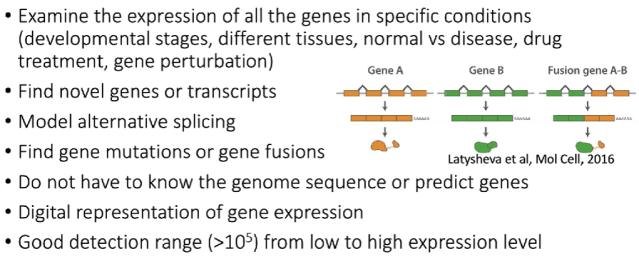
RNA-Seq的应用
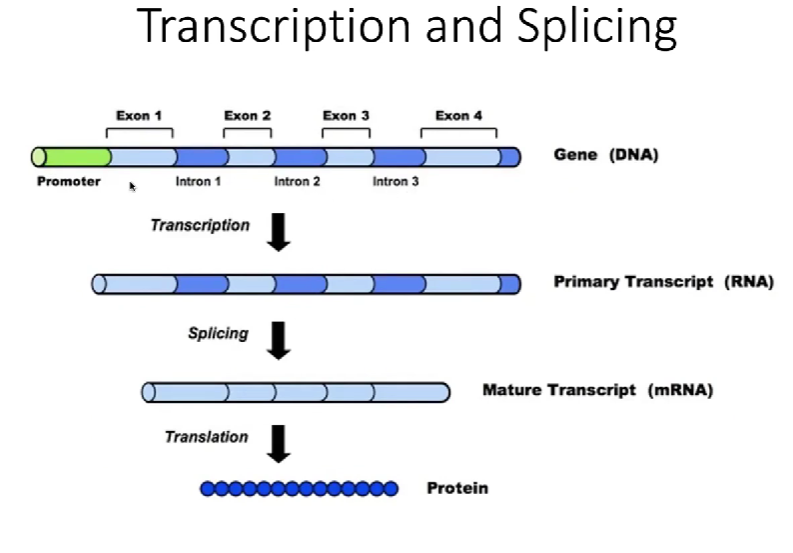
生物体内的转录&翻译过程
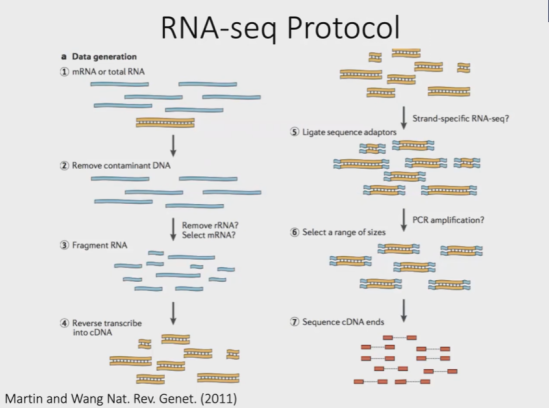
RNA-Seq建库流程
1、提取所有的mRNA或所有的RNA
2、去除DNA(在RNA建库流程中,DNA被认为是污染物)
可选过程:去除rRNA(选择mRNA)
3、将RNA片段打断(二代测序读长问题)
4、将RNA逆转录为cDNA
可选过程:链特异性选择
5、连接adapter & PCR扩增
6、选择合适长度的cDNA
7、单端测序 或 双端测序
参考文献链接:
https://www.nature.com/articles/nrg3068
RNA-Seq的应用
RNA-Seq实验设计
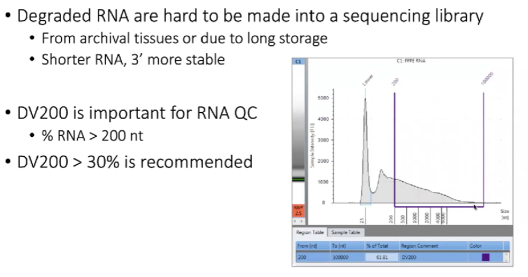
注意事项:已经降解或部分降解的RNA不可用于建库
原因:当提取的RNA被作为archival tissue储存了很长时间,同时需要注意的是短片段RNA,其3’端更加稳定。
检查RNA质量的方式:DV200指标,指片段大于200 bp的RNA的占比。
【标注】推荐DV200 > 30%
实验设计目的
RNA-Seq可以进行测序的几种RNA类型:
1、Ribo-minus:去除富集的rRNA和tRNA的RNA文库
2、使用PolyA-RNA(成熟的mRNA)进行建库
3、Strand specific/链特异性测序
【标注】一般用于挖掘novel LncRNA
测序价钱问题:一个sample 200$的情况
作为项目负责人,需要考虑的几个问题:
- SE or PE: PE getting more popular
- Depth: 20-50M for differential expression, deeper for transcript assembly or splicing.(20 million对于novel转录本和可变剪接的发现,并没有50 million的结果好)
- Read length: longer for transcript assembly, splicing, or mutation calls
4、评估biological variation
- technical replicates/技术重复:使用相同的RNA,对其测定多次;
【标注】MicroArray常用方法 - biological replicates/生物学重复:生物样本的重复
e.g. 同一株树,取3批叶片样本;患同一种癌症的人(群体)的细胞样品
【问】多少重复才足够?

RNA-Seq中的序列比对
由于在生成mRNA过程中,存在可变剪接,因此BWA等DNA序列比对软件不适用于RNA比对。
TopHat等RNA-Seq比对软件的算法,可简单概述为:
先构建参考序列的索引,将reads比对到参考基因组的exon上,同时使用不同的junction片段构建新的数据库,再将原始数据中不能比对到exon上的reads比对到junction上。
图示如下,以09年发表的TopHat作为例子:

比对生成的文件还是SAM/BAM格式,但是single-end和pair-end的数据存在一定差别:
- 第一列:read id
- 第二列:二进制标识符(如果是PE,则为奇数)
- 第三&四列:染色体编号 & 比对到染色体上的起始位置
- 第六列:cigar标识符,用于表示比对的具体情况
- 可选标识(e.g. XS,使用HISAT时可选,用于标识该序列来自正链 or 负链)
【例】

RNA-Seq:比对后的QC
RNA-Seq可能存在的问题:前几个碱基的测序质量不佳。解决方案就是使用Trimmomatic等软件将reads的前几个碱基剪切掉。
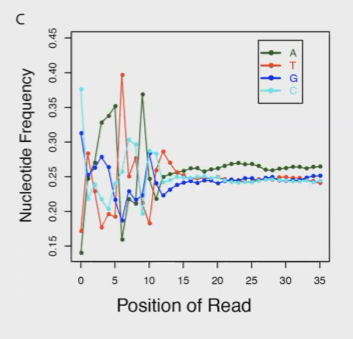
上述问题的原因,在这篇文献中有报道:https://academic.oup.com/nar/article/38/12/e131/2409775?login=true

RNA-Seq QC也可以用于查看数据的一些信息,比如插入片段长度、reads主要比对到什么区域、TIN(transcript integrity number)、medTIN(median transcript integrity number)等。如下:
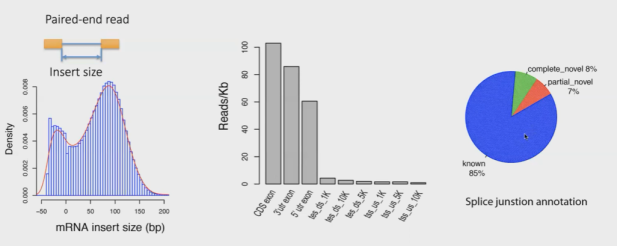
可以在这个网站查看:http://rseqc.sourceforge.net/
看这部分的时候感觉有点懵啊,不是一般都先QC再比对吗?
RPKM, FPKM, TPM
RPKM,全称“Reads Per Kilobase Million”,用于single end RNA-Seq。
计算公式:
T
o
t
a
l
r
e
a
d
s
1
M
∗
g
e
n
e
l
e
n
g
t
h
\frac{Total \quad reads}{1M * gene length}
1M∗genelengthTotalreads
FPKM,全称“Fragments Per Kilobase Million”,用于paired-end RNA-Seq
计算公式:
R
P
K
M
2
\frac{RPKM}{2}
2RPKM
TPM,全称“Transcripts Per Million”,现在最常用的RNA-Seq标准化方法。
我觉得看看这两篇文章差不多就ok了~
- 为什么都说RPKM和FPKM错了
- 浅谈RPKM, FPKM, RPM, TPM的区别
RSEM vs Salmon
(1)RSEM使用
输入数据:FASTQ or BAM
输出:转录本水平的表达量(e.g. read count, TPM, FPKM),该表达量计算结果基于转录组有效长度(effective transcript length),即exon的长度
【标注】effective length
计算公式:
l
i
−
=
l
i
−
μ
+
1
\overset{-}{l_{i}} = l_{i} - μ + 1
li−=li−μ+1
l
i
l_{i}
li为转录本长度,μ为插入片段平均长度
对于一条转录本,靠近5’端和3’端的部分较难被测序,因此若将此部分用于定量,会造成结果不准确。
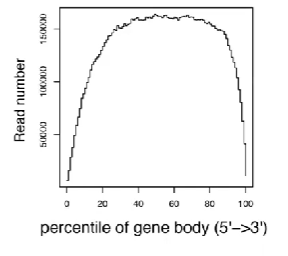
同时,还需要考虑到的因素是一个gene可以有多个isoform,如下图:
【标注】isoform,为exon组合得到的mRNA
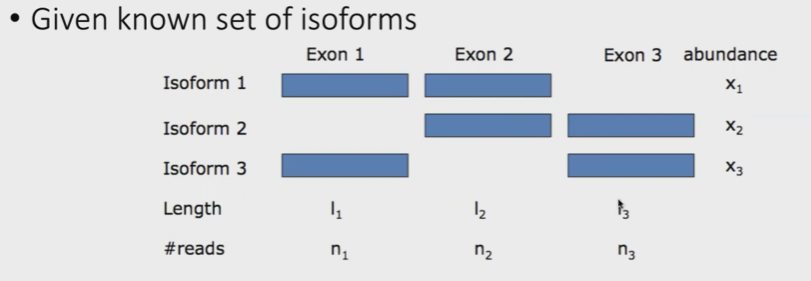
下图展示了3种不同的组织中,不同isoform的表达,同时根据reads count来估计每一种isoform在对应组织中的likelihood。
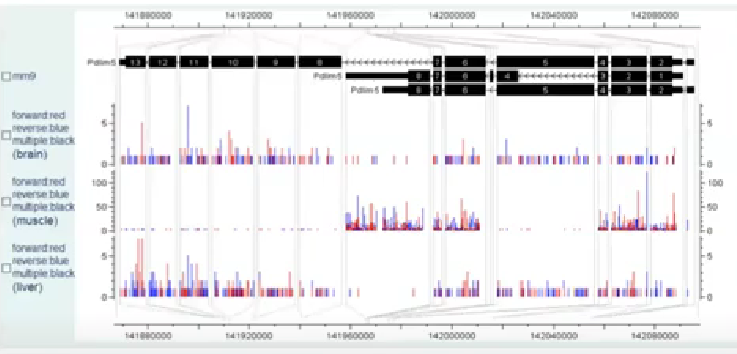
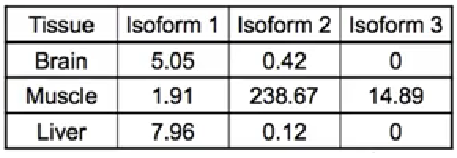
reads count表达量,如下表:
但是由于isoform3和isoform1和2,存在很多的重叠部分,因此上述分析对isoform3 likehood的估计是不准确的。
不通过比对也能够定量的方法 —— Pseudoalignment

RNA-Seq Read Distribution
(1)microarray
在芯片时代,RNA-Seq测序的reads分布,一般被认为是正态分布。
一般gene,在多个样品中的表达量分布,符合下图第三幅图:
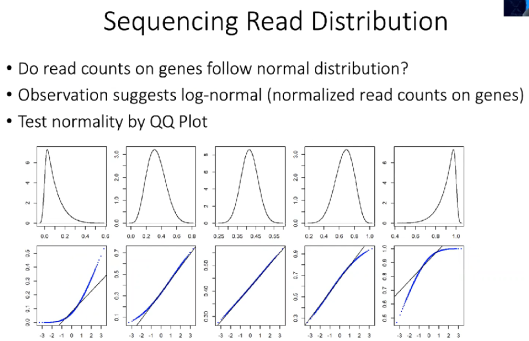
(2)RNA-Seq
RNA-Seq测序得到的reads分布,一般符合泊松分布。
e.g. 泊松分布

RNA-Seq对应的表达量 —— 负二项分布
表达量越高的gene,其对应的表达量方差越大,反之亦然。
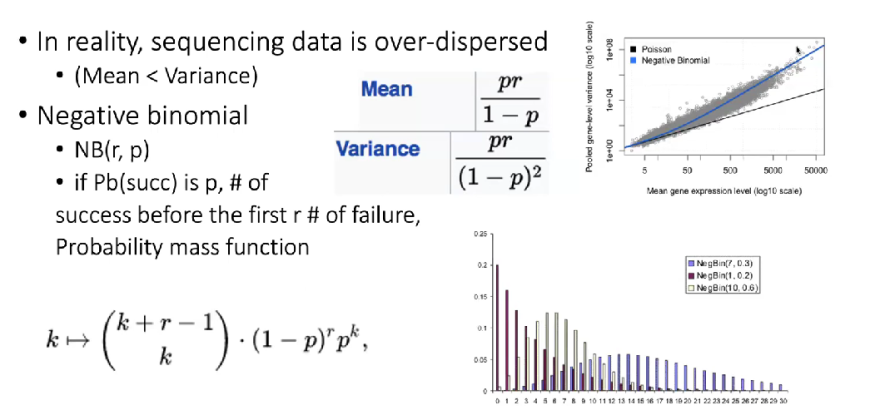
Differential Gene Expression
当获得的测序数据,不符合normal distribution(正态分布)时,可以对数据进行转换(e.g. log-transformation)
这边设计一个非常重要的概念,实际上就是针对什么类型(属于什么分布)的数据,使用什么检验方法
图示:
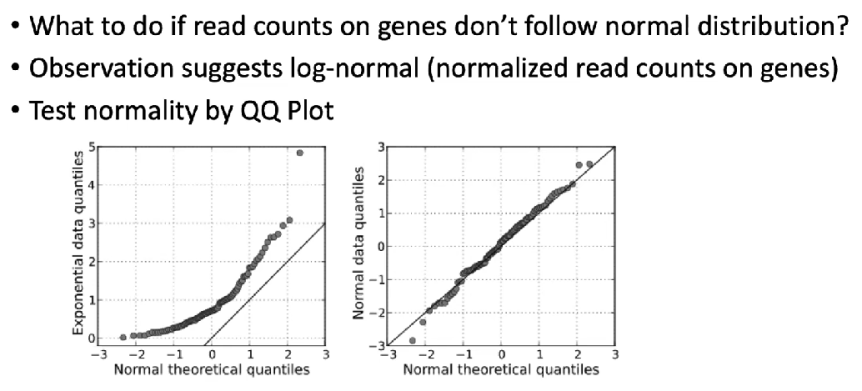
(1)microarray的标准化方式
算法:limma

【标注】一般使用t检验或t检验的变形对gene expression进行分析
limma的输入数据
(1)reads count matrix
(2)design matrix(sample属于何种实验条件)
(3)contrast matrix(对哪几种条件进行比较)
【分析标注】采用log-normal对原始数据进行转换,导致数据变泛了,对某些条件或某些gene的检测灵敏度不高了。
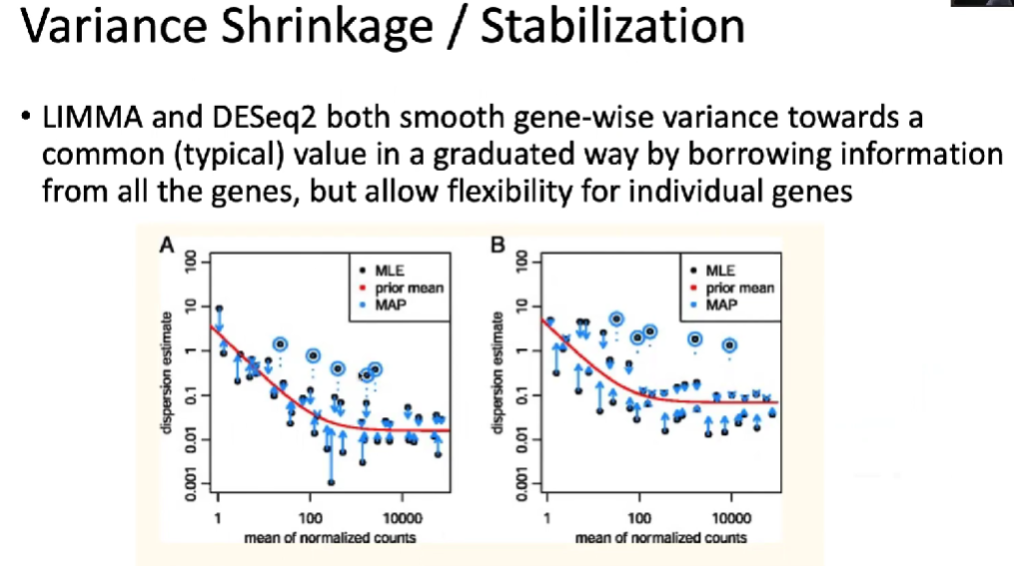
(2)基于负二项分布的标准化方法
负二项分布能够很好地捕捉到测序过程中的一些bias,对真实数据有一个更好的体现。
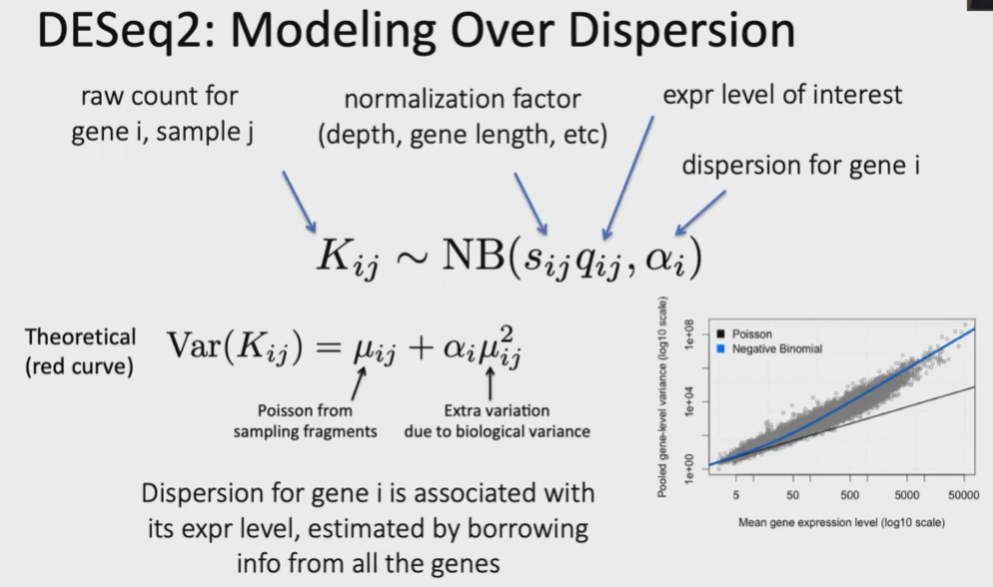
(3)DESeq2:Modeling overdispersion
RNA-Seq一般都只有2~3个rep,但是如果想要对真实的基因表达量的方差有一个体现,上述重复数肯定是不够的。
一般情况下,对基因表达量的mean和variance进行估计,估计出的方差是大于真实值的,如下图蓝线和黑线的区别:
差异表达基因的可视化
(1)火山图
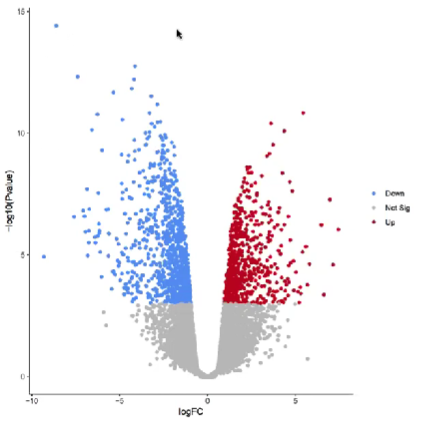
(2)MA图
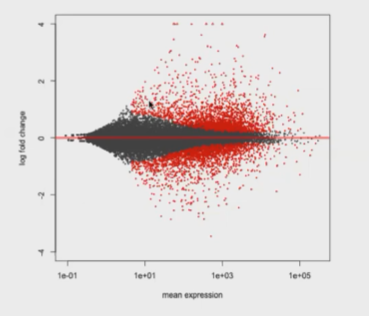
M:代表两种实验条件下的log ratio和log fold change
A:基因表达量的均值
后话
虽然跑过转录组,但是实际上我对其中的原理只能说是一知半解,还需要多实践。
课程得学,笔记也得做,实践也得跟上。
但是觉得笔记还是不够硬核,或许应该给自己看看就好。















 498
498











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









