









简介
blast是常用的比对软件,在linux系统下安装完成blast套件后,可以使用blastn进行核酸序列的比对,基本的使用模式为确定搜索的库,然后使用blastn对指定的序列在库中进行比对,如果想要自定义搜索库,需要使用makeblastdb来创建,更多的细节可以参考blast 官方文档接下来我将介绍如何创建自定义搜索库并进行搜索的步骤。
使用makeblastdb创建自定义搜索库
创建自定义搜索库需要事先准备号一个fasta文件,fasta文件中存在用于创建搜索库的所需序列,形式如下图所示:
接下来使用该fasta文件创建库文件
makeblastdb \
-dbtype nucl \
-in test.fasta \
-input_type fasta \
-parse_seqids \
-out test.blastdb
参数说明:
-dbtype 是必选参数,nucl, prot,二选一,核酸序列或者氨基酸序列库
-in 用于构建搜索库的fasta文件
-input_type 默认为fasta文件,其他支持文件asn1_bin,asn1_txt, blastdb
-parse_seqids 添加该参数,结果文件将多三个文件,不加默认为3个结果文件
-out输出文件的前缀
执行上述代码后将生成后缀为nin,nhr,nsq,nsi,nsd,nog六个文件,至此,自定义搜索库文件生成完成
blastn
在生成自定义搜索库文件之后就可以使用该库文件进行搜索了,搜索代码如下:
blastn \
-query test.fasta \
-db test.blastdb \
-out blastn_results.xls \
-task blastn
参数说明
-query 指定输入文件
-db 指定搜索数据库,这里只需要输入前缀就可以了
-out输出文件名称
-task 搜索算法,默认为blastn,不需要添加,其他可选参数为blastn,blastn-short,dc-megablast,megablast,rmblastn
执行完成后得到如下形式的输出结果
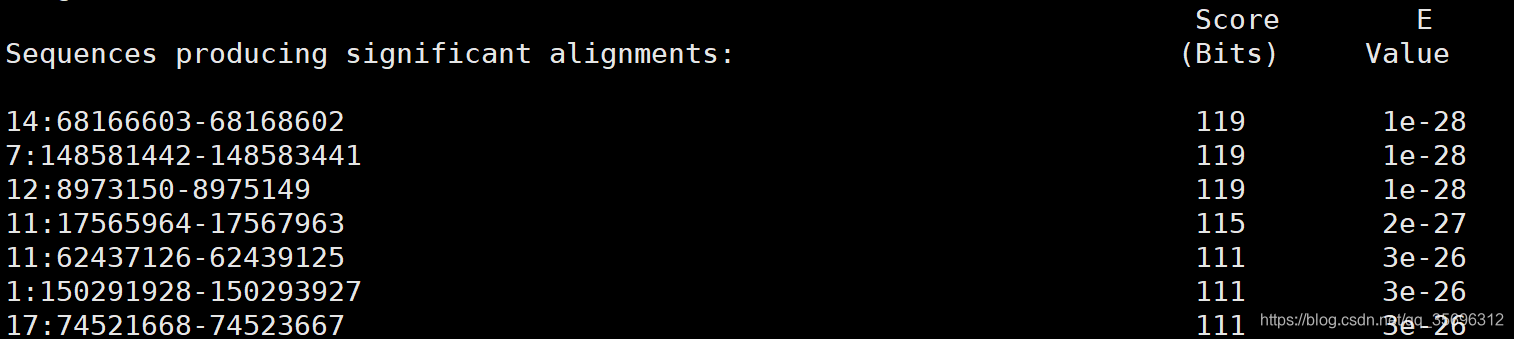
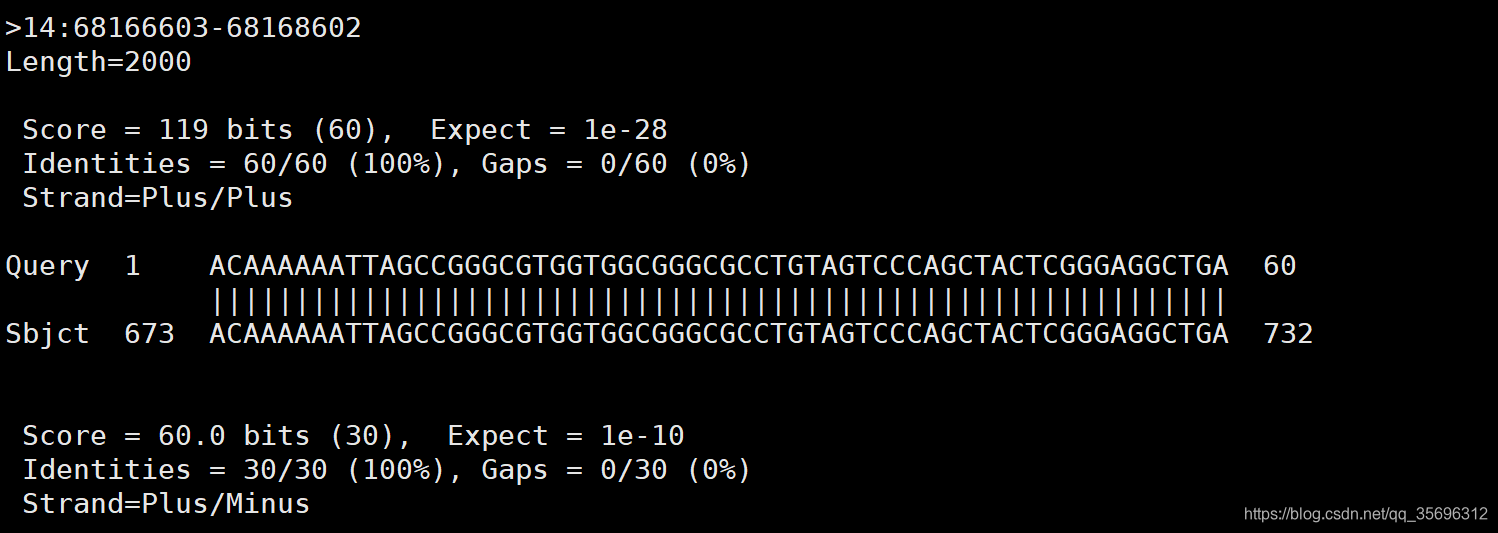
包括了比对的位置以及得分和E值,E值越低说明这种比对特异性越强,越可信。除此以外,文件的下面还有详细的比对情况:
极短序列比对
对于特别短的序列例如20bp左右的序列,在查找的时候我们需要添加三个参数,如下代表所示:
-task blastn-short
-word_size 4 \
-evalue 1 \
-task参数指定为短序列的匹配算法
-word_size该参数只能指定4以上的值,这里写了最小的4,指定用于匹配算法的最短序列
-evalue 该值越小,得到的结果将更加严苛,这里设定为1即可。

















 8343
8343

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









