







之前在平台发布了几个问题,接下来我会依次给出我的想法,仅供参考。本文有些观点读者朋友可能是第一次看到,有不同意见的话,欢迎给我发邮件讨论:huangsiyuan1001@163.com
问题
- 一、有的文章只提供TPM的单细胞表达矩阵,可以用seurat分析吗?
- 二、分析流程和用count矩阵有什么不同?
- 三、10X的单细胞转录组数据的标准化需要考虑基因长度吗?
先来看看第3个小问题
10X的单细胞转录组数据的标准化需要考虑基因长度吗?
答案是不需要。
我们看一下seurat里面NormalizeData()函数是如何做标准化,然后求Log。
test.seu <- NormalizeData(test.seu, normalization.method = "LogNormalize", scale.factor = 10000)
LogNormalize: Feature counts for each cell are divided by the total counts for that cell and multiplied by the scale.factor(默认是10000). This is then natural-log transformed using log1p.(每一列都是先除以一个细胞UMI的总和,再乘以10000,再加1,再求对数)
的确没有考虑基因长度。
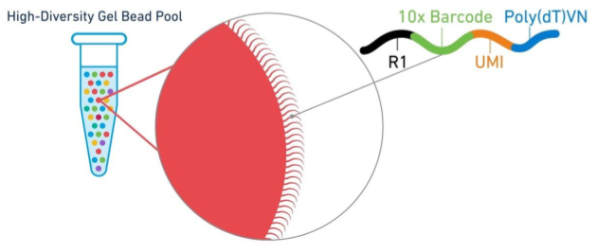
这要从测序原理去解释。以10X的3’端测序为例,磁珠上有很多短序列(它们的cellular barcode是一样的,UMI各不相同),在PolyT结合mRNA的3’端PolyA之后,尽管转录本有长有短,但因为测序序列长度有所限制,这些转录本分子会在靠近3’端的地方随机打断(只留下最靠边的序列),
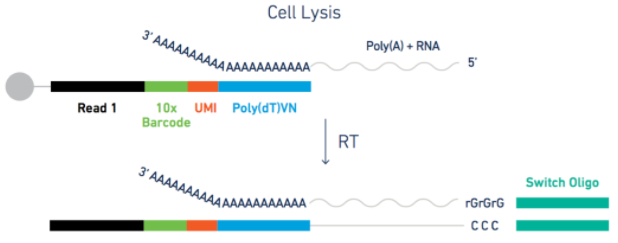
然后两边连上接头进行后续的测序步骤…
所以表达定量的多少和基因长度关系不大。另外,多说一句,UMI的作用是消除PCR的影响,只要是来源于一个转录本,不管扩增多少次,最后定量值只会加1。
那传统二代转录组测序为什么考虑基因长度呢?
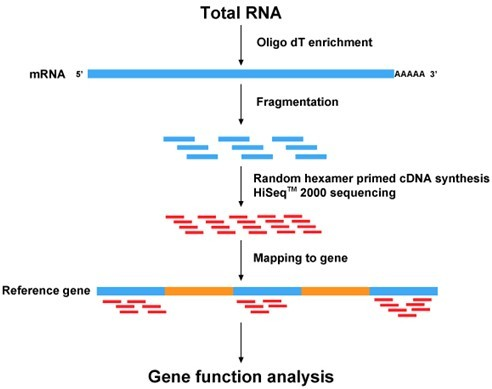
还是从测序原理去解释。一个完整的转录本分子会被随机打断,转录本越长,片段会越多,而这些片段最终都会被测序。因为这个偏好,标准化的时候才需要考虑转录本长度的影响。
回到第1个小问题
TPM的单细胞表达矩阵,可以用seurat分析吗?(可以)
先来看一下TPM的公式,顺带把RPKM的也看了:

上述公式把转录本长度这一项去掉,就会发现和前面讲的seurat的LogNormalize方法中的标准化这一步是如此类似
“每一列都是先除以一个细胞UMI的总和,再乘以10000”
只是乘上的倍数不同,形式是一样的。把umi/sum(umi) * 10000当作TPM、RPKM的特例。既然如此,TPM、RPKM这一类标准化形式的数据应该也是能走seurat标准流程的,只要把文库大小、转录本长度这些因素考虑了就行。代码略有不同,待会儿再说。
类似的问题,比如从10X单细胞数据中导出类似TPM的矩阵,是不是也很简单了呢?从seurat的count矩阵和data矩阵都能推算出来。
为啥会问这个问题?之前有读者问我运行cibersort的时候,需要导入单细胞得到的参考矩阵,需要类似TPM的数据(要和bulk数据保持一致),行是基因,列是细胞类型。
在得到类TPM矩阵后,手动计算每种细胞类型的均值,当然也可以用seurat的AverageExpression这个函数,结果一样。
### 10X导出类tpm矩阵 #####
### 第一种方法
test.seu=readRDS("D:/hsy/bioinformatics/PTJ/011_paper/CR/malignant.seu.1129.rds")
mat.tpm.like=as.data.frame(test.seu[["RNA"]]@counts) %>% apply(2,function(x){x/sum(x) * 10000})
#减少计算量,只选择一个亚群
selectCB=test.seu@meta.data[test.seu@meta.data$sample == "P14","CB"]
mat.tpm.like=mat.tpm.like[,selectCB]
#手动计算均值
selectCB.tpm.mean=as.data.frame(rowMeans(mat.tpm.like))
colnames(selectCB.tpm.mean)="method1_P14"
### 第二种方法
Idents(test.seu)="sample"
averageexp=AverageExpression(test.seu)
averageexp=averageexp$RNA
averageexp=as.data.frame(averageexp[,"P14"])
colnames(averageexp)="method2_P14"
### 比较两种方法
twomethod=cbind(averageexp,selectCB.tpm.mean)
twomethod$d=twomethod$method2_P14-twomethod$method1_P14
twomethod$d=round(twomethod$d,5)
> table(twomethod$d)
0
33538
# 差值都是0,说明两种方法求出来是一样的
相信你现在对AverageExpression函数有了一个更深的理解,现在知道它返回的是什么了吧。
我想这也是为什么早期有几篇文章描述10X的数据会用到TPM这个词:
Our data were transformed into log 2 (TPM+1), where transcripts per million
(TPM) was calculated as the proportion of UMIs of a gene within
each cell multiplied by 1,000,000.
(来自文献: Single-cell transcriptomics reveals regulators underlying
immune cell diversity and immune subtypes associated with
prognosis in nasopharyngeal carcinoma)
OK, 上述都是我个人的理解。seurat官方GitHub是这么说的:
- If you have TPM data, you can simply manually log transform the gene expression matrix in the object@data slot before scaling the data.
- You have to replace your object@data slot with the desired gene expression matrix as follows: pbmc@data = log(x = norm + 1))
(分别来自https://github.com/satijalab/seurat/issues/668和https://github.com/satijalab/seurat/issues/747)
第2个小问题
在分析流程上,这种矩阵和用count矩阵有什么不同?
只有前几步有点区别,
### 常规流程
test.seu=CreateSeuratObject(counts = testdf)
test.seu <- NormalizeData(test.seu, normalization.method = "LogNormalize", scale.factor = 10000)
test.seu <- FindVariableFeatures(test.seu, selection.method = "vst", nfeatures = 2000)
......
### TPM数据
test.seu1=CreateSeuratObject(counts = log(mat_tpm+1))
test.seu1 <- FindVariableFeatures(test.seu1, selection.method = "mvp", nfeatures = 2000)
# 或者
test.seu=CreateSeuratObject(counts = mat_tpm)
test.seu[["RNA"]]@data=as(as.matrix(log(mat_tpm + 1)), "dgCMatrix")
test.seu <- FindVariableFeatures(test.seu, selection.method = "mvp", nfeatures = 2000)
......
后面的流程一样。有人会担心,提供TPM,不能找差异基因。事实上,seurat默认找差异基因的方法并没有用count,而是data矩阵,所以还是可以用FindMarkers()的。
本文图片来自谷歌和10X官网;本文有些观点各位读者朋友大概率是第一次看到,仅代表我个人观点。
欢迎点赞、转发、转载!















 6641
6641











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









