









是一篇来自2018年10月7日的笔记。当时做了一批土壤氨氧化细菌(AOA/AOB)的amoA基因的克隆测序,需要对测序结果进行批量鉴定。为了节省时间,自学了blast本地比对,当时还使用的blast-2.7.1+版本。现在随意从NCBI下载了一些序列,重现并更新一下笔记。
一、软件安装
现在是ncbi-blast-2.12.0+版本,下载网址:ftp://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/LATEST/。
# Linux下载
wget -c -t 0 ftp://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/LATEST/ncbi-blast-2
.12.0+-x64-linux.tar.gz
# 也可下载windows版本使用
ftp://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/LATEST/ncbi-blast-2.12.0+-win64.exe
# 解压缩
tar -zxvf ncbi-blast-2.12.0+-x64-linux.tar.gz
# 将blast的子程序所在bin目录设置为环境变量
echo "export PATH=~/hj/software/ncbi-blast-2.12.0+/bin/:\${PATH}" >> ${HOME}/.bashrc
source ~/.bashrc # 或export PATH=~/hj/software/ncbi-blast-2.12.0+/bin/:${PATH}

图1|blast包含的子程序。软件使用手册:http://www.ncbi.nlm.nih.gov/books/NBK279690。
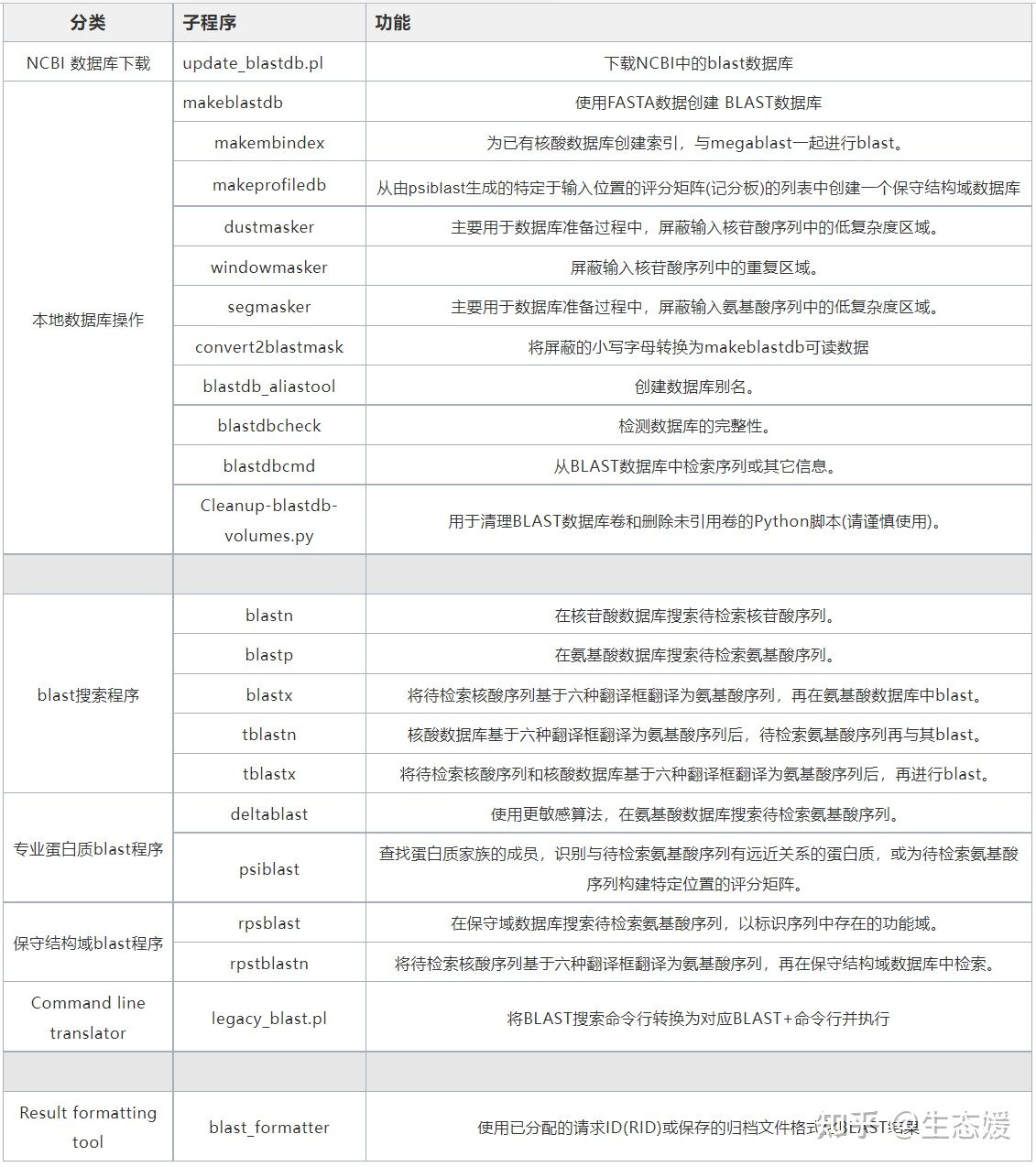
表1|blast子程序使用说明(https://www.ncbi.nlm.nih.gov/books/NBK52640/)
二、数据准备
NCBI用于注释的数据库,可以使用研究者自己的FASTA序列使用makeblastdb创建。也可使用NCBI现成blast数据库,解压直接使用。blast数据库下载地址:https://ftp.ncbi.nlm.nih.gov/blast/db/。数据库信息说明网站:https://www.ncbi.nlm.nih.gov/books/NBK62345/#blast_ftp_site.The_blastdb_subdirectory。GENOME TAXONOMY DATABASE(https://gtdb.ecogenomic.org/)是细菌和古菌基因组数据库。Fungene(http://fungene.cme.msu.edu/hmm_detail.spr?hmm_id=358)可以下载功能基因序列,目前因为硬件故障不能下载了,不知道什么时候才能好。还有很多其它的数据库,可以自行去了解。
表2|部分blast数据库。数据库有多个子文件的,需要一起下载。查看数据库对应metadata.json文件,可以查看数据库版本、数据库类型和包含数据文件等信息。也可在国家微生物科学数据中心的备份链接(ftp://download.nmdc.cn/blast/db/)中下载,但是更新有延迟。RefSeq Select数据库包含每个蛋白编码基因的的代表性和可选择序列(https://www.ncbi.nlm.nih.gov/refseq/refseq_select/)。标记基因项目:https://www.ncbi.nlm.nih.gov/refseq/targetedloci/。
2.1 下载blast v5数据库
# 下载、更新blastv5数据库
## 自行下载解压缩
cd ~/hj/DB
nohup wget -t 0 -c https://ftp.ncbi.nlm.nih.gov/blast/db/16S_ribosomal_RNA.tar.gz >16s.log 2>&1 &
nohup wget -t 0 -c https://ftp.ncbi.nlm.nih.gov/blast/db/18S_fungal_sequences.tar.gz >18s.log 2>&1 & # 下载数据库
nohup wget -t 0 -c https://ftp.ncbi.nlm.nih.gov/blast/db/ITS_RefSeq_Fungi.tar.gz >ITS_F.log 2>&1 &
nohup wget -t 0 -c https://ftp.ncbi.nlm.nih.gov/blast/db/ITS_eukaryote_sequences.tar.gz >ITS_E.log 2>&1 &
nohup wget -t 0 -c https://ftp.ncbi.nlm.nih.gov/blast/db/cdd_delta.tar.gz >CDD.log 2>&1 &
for file in `find . -name "*.gz"`
do
fold=`basename ${file} .tar.gz`
mkdir "$fold";
tar zxvf "$file" -C "$fold";
done # 批量解压缩blast数据库到指定目录中,防止taxdb信息被覆盖。## 使用update_blastdb.pl下载或更新数据库
update_blastdb.pl --showall # 查看所有数据库
### 未设置blast环境变量,则使用perl脚本的全路径运行程序
perl ~/software/ncbi-blast-2.12.0+/bin/update_blastdb.pl --passive \
--decompress taxdb \
--blastdb_version 5 # 若taxdb数据库已存在,则此步骤为更新taxdb数据库。
###update_blastdb.pl有时会报错,手动下载更便捷。
## 设置数据库BLASTDB环境变量-为了提高使用效率(https://www.ncbi.nlm.nih.gov/books/NBK52640/)
echo "export BLASTDB=~/hj/DB" >> ~/.bashrc
#### 未设置BLASTDB,blast将会搜索工作目录## 检查数据库的完整和有效性,# pal是protein后缀;nal是核酸后缀
~/software/ncbi-blast-2.12.0+/bin/blastdbcheck -help # 查看帮助信息
### 检测1个指定数据库,-full表示检测数据库中的每一条序列,数据量很大可以换成 -random,-ends或 -stride
~/software/ncbi-blast-2.12.0+/bin/blastdbcheck \
-db /home/zhangguogang/hj/DB/16S_ribosomal_RNA/16S_ribosomal_RNA \
-dbtype nucl -verbosity 3 -full
### 递归检测目录下所有数据库的完整性,-verbosity设置检测结果的输出详细程度0-4可选
~/software/ncbi-blast-2.12.0+/bin/blastdbcheck \
-dir ~/hj/DB/ \
-recursive \
-dbtype guess \
-verbosity 2 \
-full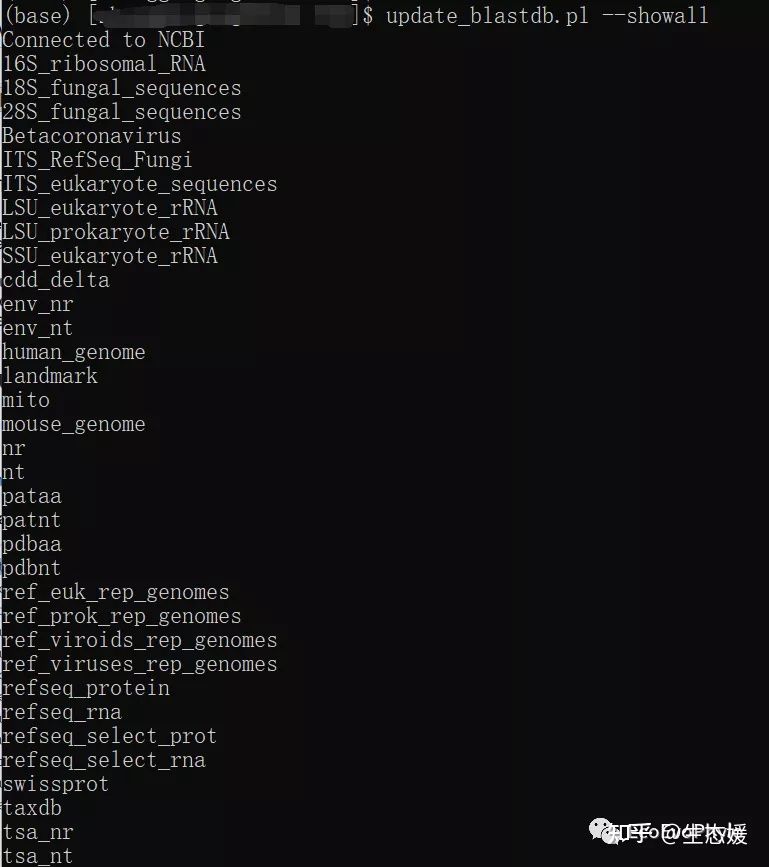
图2|update_blastdb.pl可更新下载数据库名称。
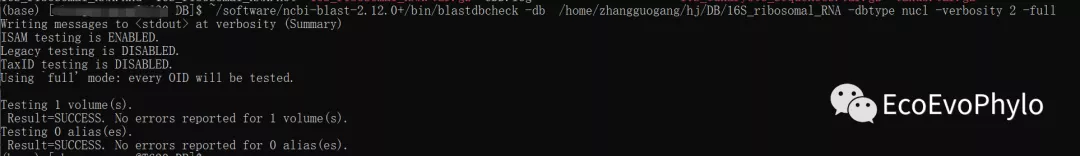
图3|blastdbcheck检测数据库完整性。
2.2 用于blast比对的序列准备
当时因为做的是AOA/AOB-amoA基因的克隆测序,所以我下载了NCBI中的所有AOA/AOB-amoA基因的fasta格式序列,用makeblastdb构建数据库,然后再进行比对。这里就使用blastdbcmd下载一些序列用于学习blast操作。也可以从NCBI(可以使用edirect本地下载)或其他数据库下载DNA或protein序列,最好有GI号。
# blast v5数据的可使用LMDB(http://www.lmdb.tech/doc/)
##或blastdbcmd通过登录号更快速的检索序列
blastdbcmd -help #查看帮助信息
## 使用登录号本地下载序列,
###也可以在http://www.ncbi.nlm.nih.gov/sites/batchentrez网址手动下载
cd ~/hj/blast
### 下载一个登录号序列
~/software/ncbi-blast-2.12.0+/bin/blastdbcmd \
-db $BLASTDB/16S_ribosomal_RNA/16S_ribosomal_RNA \
-entry nr_025000 -out 16S_query.fa
### 批量下载多个登录号序列
cat batch_Accessions.txt # batch_Accessions.txt中包含3个登录号
nr_025000
nr_028940
nr_125568
~/software/ncbi-blast-2.12.0+/bin/blastdbcmd \
-db $BLASTDB/16S_ribosomal_RNA/16S_ribosomal_RNA \
-entry_batch batch_Accessions.txt -out 16S_query3.fa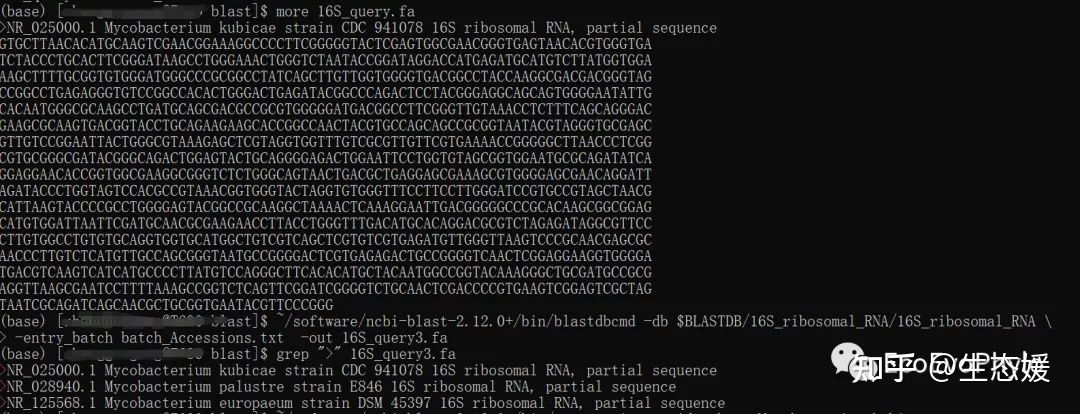
图4|blastdbcmd根据登录号从blast数据库中获取序列。
## 可以使用taxid从NCBI下载数据,
###taxid是NCBI为各分类单元赋予的独一无二的数字ID。
###可以在taxonomy数据库(https://www.ncbi.nlm.nih.gov/Taxonomy/Browser/wwwtax.cgi)
###或使用blast中的get_species_taxids.sh基于拉丁或科学名称查询
### 获取taxid列表
~/software/ncbi-blast-2.12.0+/bin/get_species_taxids.sh \
-n Mycobacterium kubicae # taxid是属分类单元1763
~/software/ncbi-blast-2.12.0+/bin/get_species_taxids.sh \
-t 1763 > 1763.txids # 获取1763分类等级及之下的所有taxid
### 使用taxid列表下载数据,不是所有taxid都能从数据库中下载到数据。
~/software/ncbi-blast-2.12.0+/bin/blastdbcmd \
-db $BLASTDB/16S_ribosomal_RNA/16S_ribosomal_RNA \
-taxidlist 1763.txids \
-outfmt "%f %T %S" \
-target_only \
-out 16S_query_MK.fa
#### 使用taxid下载序列,1233042表示ammonia-oxidizing archaeon AR。
####输出格式中%a表示输出登录号, %T输出taxid, %S输出科学名称。
#### -target_only # 因为nr是一个非冗余数据库,一个序列可能对应多个登录号。
####设置-target_only表示只返回结果中只包含待检索登录号的序列,
####否则只要至少包含一个待检索登陆号的序列都会被返回。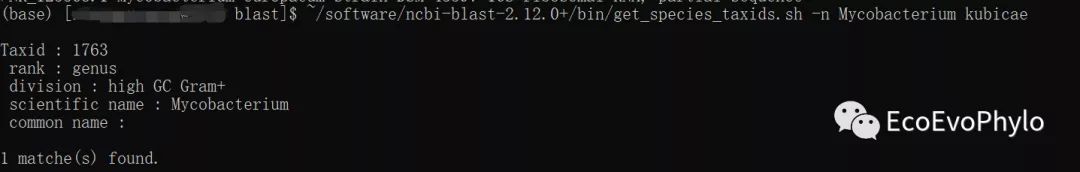
图5|get_species_taxids.sh获取物种分类单元ID。
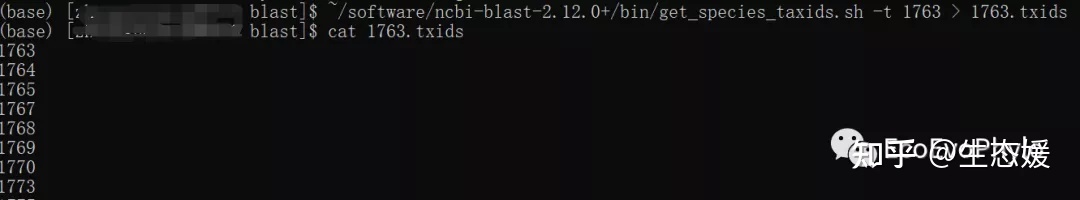
图6|get_species_taxids.sh获取某分类单元下的所有taxid。

图7|blastdbcmd使用taxid列表下载序列数据。
也可手动再NCBI batchentrez网站(http://www.ncbi.nlm.nih.gov/sites/batchentrez)下载:1.将需要下载的序列的序列号整理为一个txt文件,一行一个2.NCBI批量下载序列网址上传txt文件下载
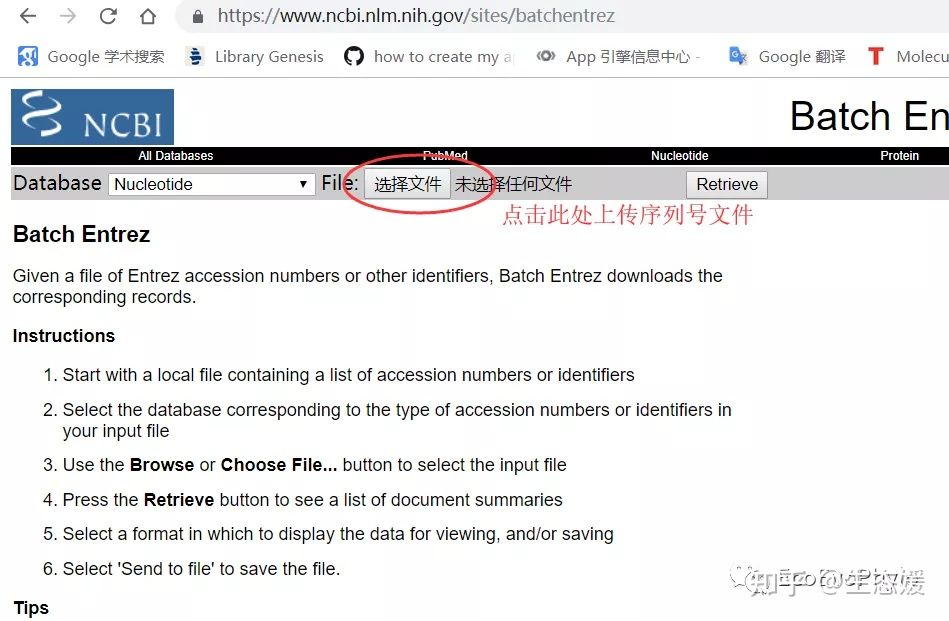
三、blast本地比对流程-blastn为例
准备好数据库和需要比对的序列,就可以使用blast的搜索子程序(blastn,blastp,blastx,tblastn和tblastx)进行序列检索。windows和Linux中都能运行。blast使用手册:https://www.ncbi.nlm.nih.gov/books/NBK279690/
3.1 使用下载的blast数据库进行序列检索
# 3.1 使用下载的blast数据库进行序列检索
## 3.1.1 使用blastn(核酸to核酸),在16S RNA数据库中检索16S_query3.fa序列
blastn -help # 帮助信息
#blastn 比对类型,打开输出文件查看比对结果
~/software/ncbi-blast-2.12.0+/bin/blastn \
-db $BLASTDB/16S_ribosomal_RNA/16S_ribosomal_RNA \
-query 16S_query3.fa \
-task blastn \
-strand both \
-evalue 1e-5 \
-max_target_seqs 1 \
-outfmt 6 \
-out 16S_blast_out
## -strand both:可以选择序列检索方向:both, minus或plus。
## -task:设置检索方式,默认`megablast,
###一般选择blastn,待检索序列碱基数<50,选择blastn-short进行检索。
## -max_target_seqs 1:显示匹配度最高的一条序列
## -outfmt 6 #有17种格式可选,
###很多基因结构和功能注释都是基于blast(dimond现在使用的也很多,运行更快)比对的输出结果,
###常用XML(5)和 Tabular(6,7)等格式,
###输出结果 6, 7和10形式,可以自己添加额外的输出参数。
表3|blastn各搜索选项。https://www.ncbi.nlm.nih.gov/books/NBK569839/#usrman_BLAST_feat.Sequence_filtering_app
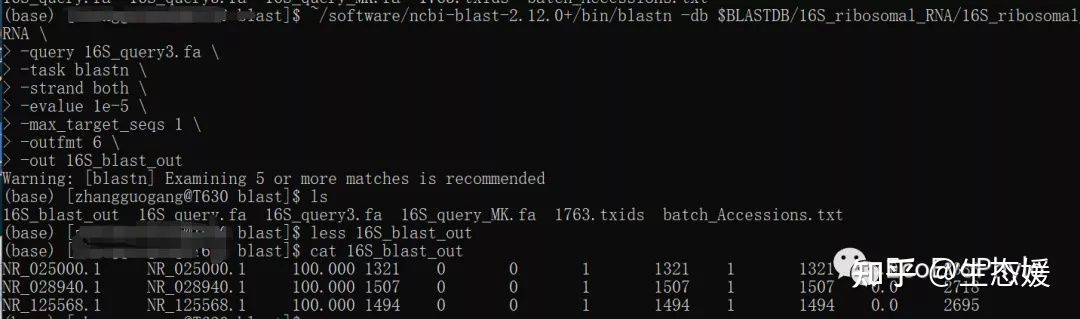
图8|blastn的outfmt 6的输出结果。16S_blast_out。
## 3.1.2 自定义输出内容:为了不检索到待检索序列本身,
###将待检索序序列ID整理为rm.txt文档,
###检索时使用-negative_seqidlist用于排除序列id
grep ">" 16S_query3.fa | awk '{print $1}' | sed 's/>//g' > rm.txt
~/software/ncbi-blast-2.12.0+/bin/blastn \
-db $BLASTDB/16S_ribosomal_RNA/16S_ribosomal_RNA \
-query 16S_query3.fa \
-task blastn \
-strand both \
-evalue 1e-5 \
-negative_seqidlist rm.txt \
-num_descriptions 5 \
-num_alignments 5 \
-outfmt "7 std staxid sstrand" \
-num_threads 4 \
-out 16S_blast_out2
# 输出结果在7的标准(std)格式下,
##增加一列分类单元ID(staxid)和一列检索到的序列的链方向。
## -num_descriptions 500 #要显示单行说明的数据库序列数,
###与-max_target_seqs不兼容, outfmt > 4就不能设定了。
## -num_alignments 250 #输出结果中包含比对到的序列的数量,
###与-max_target_seqs不兼容
## -num_threads 4 # 设置检索是使用的CPU数量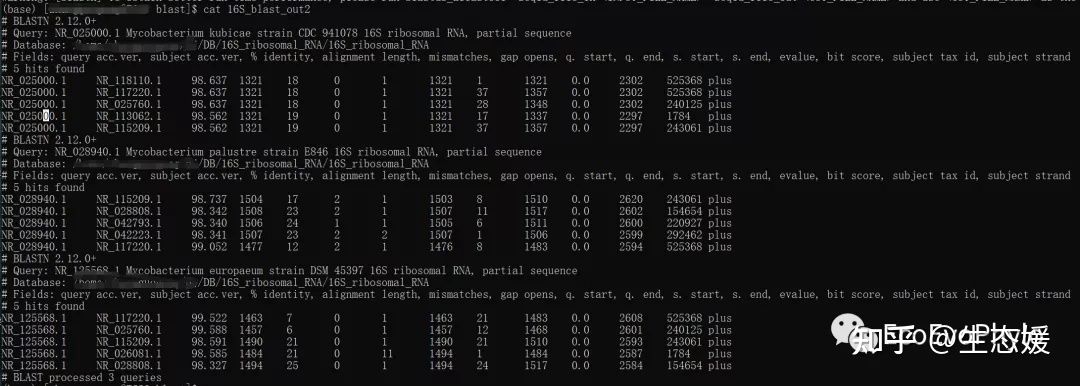
图9|blastn的outfmt 7加分类单元和链方向的输出结果。 16S_blast_out2比对结果不包含它们自身了。
## 3.1.3 如果需要屏蔽的数据库序列很多或者想在数据库指定序列中进行比对
### 可以使用blastdb_aliastool根据序列ID生成bsl文件,
###用于指定序列,加快检索速度。
~/software/ncbi-blast-2.12.0+/bin/blastdb_aliastool \
-seqid_file_in rm.txt -seqid_file_out rm.txt.bsl
~/software/ncbi-blast-2.12.0+/bin/blastn \
-db $BLASTDB/16S_ribosomal_RNA/16S_ribosomal_RNA \
-query 16S_query3.fa \
-task blastn \
-evalue 1e-5 \
-seqidlist rm.txt.bsl \
-outfmt "17 std SQ" \
-out 16S_blast_out3
#### -seqidlist rm.txt.bsl,与指定序列进行比对,-
#### outfmt "17 std SQ"输出碱基序列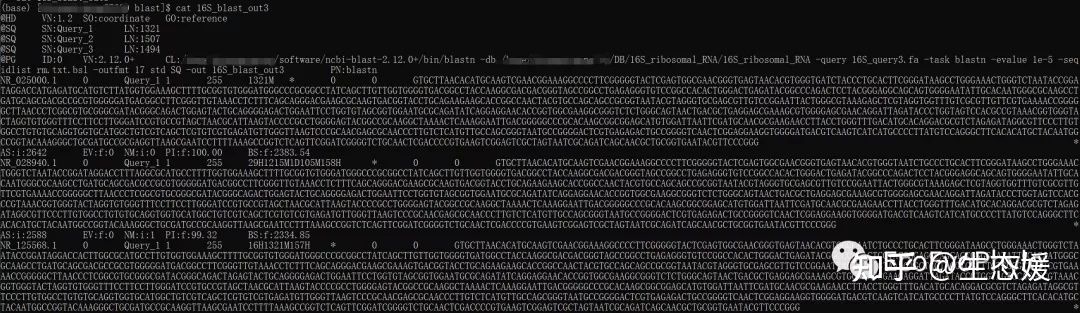
图10|blastn的outfmt 17加碱基序列的输出结果。16S_blast_out3比对结果只包含自身。
## 3.1.4 比对时,也可以指定分类单元ID进行比对或屏蔽特定分类单元ID
###取指定物种分类单元对应的taxid
#/software/ncbi-blast-2.12.0+/bin/get_species_taxids.sh -n Mycobacterium kubicae # taxid是属分类单元1763
#/software/ncbi-blast-2.12.0+/bin/get_species_taxids.sh -t 1763 > 1763.txids # 获取1763分类等级及之下的所有taxid
### 指定分类单元ID进行比对或屏蔽特定分类单元ID
~/software/ncbi-blast-2.12.0+/bin/blastn \
-db $BLASTDB/16S_ribosomal_RNA/16S_ribosomal_RNA \
-query 16S_query3.fa \
-taxidlist 1763.txids \
-outfmt "6 std staxid" \
-out 16S_blast_out5 # –taxidlist 1763.txids # 可指定多个txid
##-negative_taxidlist选项用于排除对应选项
~/software/ncbi-blast-2.12.0+/bin/blastn \
-db $BLASTDB/16S_ribosomal_RNA/16S_ribosomal_RNA \
-query 16S_query3.fa \
-taxids 1273442 \
-outfmt "7 std staxid" \
-out 16S_blast_out6 # -taxids 1273442 #需要物种及更低分类水平的taxid
## -negative_taxids用于排除对应选项
## -query_loc:可以选择待查询序列的指定序列位置进行检索,比如-query_loc 102-1110,表示待检索序列的第102个碱基到1110碱基。
## NCBI网页版blast上能设置的参数,blast本地版都能设置,查看帮助信息设置即可。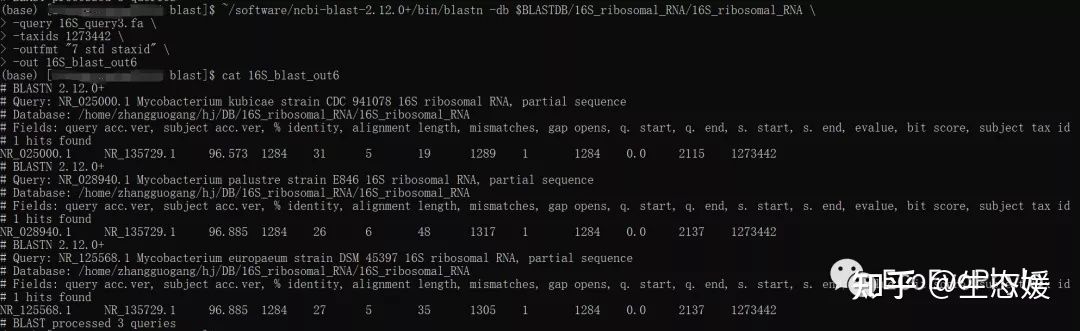
图11|-taxids 1273442的输出结果。16S_blast_out6只包含1273442分类单元ID序列。
3.2 使用其他数据库下载或测序得到的FASTA序列构建数据库进行blast比对
# 使用makeblastdb构建数据库
## chmod 777 ~/software/ncbi-blast-2.12.0+/bin/makeblastdb # 赋予运行权限
~/software/ncbi-blast-2.12.0+/bin/makeblastdb -help # 查看帮助信息
~/software/ncbi-blast-2.12.0+/bin/makeblastdb -in 16S_query3.fa \
-dbtype nucl -out 16S_query3db -parse_seqids
## -input_type fasta #用于构建数据库的数据形式有4种可选:asn1_bin, asn1_txt, blastdb和fasta(默认)
## -dbtype nucl # 构建的数据库类型nucl:核酸数据库;prot:蛋白数据库。
## -out 16S_query3db #数据库命名,会输出以16S_query3db为前缀的一系列数据库文件。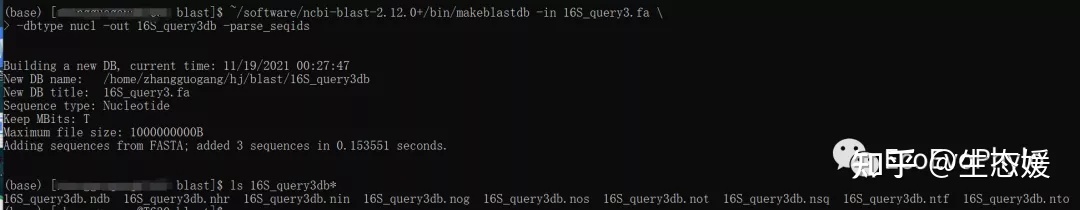
图12|数据库构建结果。16S_query3db
# 数据库构建完成,就可以使用blastn进行检索了
#blastn -help # 帮助信息
~/software/ncbi-blast-2.12.0+/bin/blastn -db 16S_query3db \
-query 16S_query3.fa \
-task blastn \
-evalue 1e-5 \
-outfmt 6 \
-out 16S_blast_out4
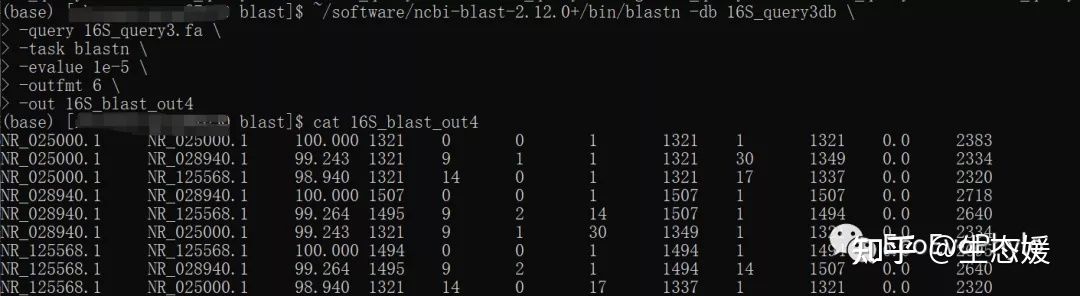
图13|自建数据库检索结果。数据库路径要正确。16S_blast_out4
# windows下使用方法是一样的,进入程序所在位置
#进入blast单机版所在磁盘
H:
#进入bin文件夹,bin文件夹下有多种可运行程序
cd H:\software\NCBI\\bin
#新建比对序列数据库,出现数据库文件namedb.nhr/nin/nsq
makeblastdb -in 序列文件名.fasta -dbtype 数据库类型 -out 输出数据库名db -parse_seqids
#可能会出现将无法将命令识别为程序名,则在命令行前添加“.\”
.\makeblastdb -in 序列文件名.fasta -dbtype 数据库类型 -out 输出数据库名dbblast其他子程序的使用大同小异,查看帮助信息和使用手册使用即可。blast也可对多重比对序列(https://www.ncbi.nlm.nih.gov/books/NBK569842/)进行检索。rpsblas可用于基因家族分析时进行结构域搜索。blast的功能很多,很强大,有需要可以看一下blast的使用手册。
微信公众号后台发送"blast",可以获取软件包和说明文档,或在QQ交流群中的文件夹中下载。
推荐阅读
IsoformSwitchAnalyzeR-基于转录本的可变剪切及isoform Switch分析
Phylogenomics-构建不分区串联全基因组进化树(ML法)
Phylogenomics-序列比对multi-FASTA转为VCF及VCF版本转换
EcoEvoPhylo :主要分享微生物生态和phylogenomics的数据分析教程。
扫描二维码,关注EcoEvoPhylo。让我们大家一起学习,互相交流,共同进步。
学术交流QQ群 | 438942456
学术交流微信群 | jingmorensheng412
加好友时,请备注姓名-单位-研究方向。


















 3万+
3万+

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











