










前情回顾
OmicsTools的安装使用和R语言分析环境的配置教程
 https://blog.csdn.net/qq_40073899/article/details/139143993
https://blog.csdn.net/qq_40073899/article/details/139143993根据GSE id自动下载处理GEO数据(必须要运行的模块)
该模块的运行窗口截图
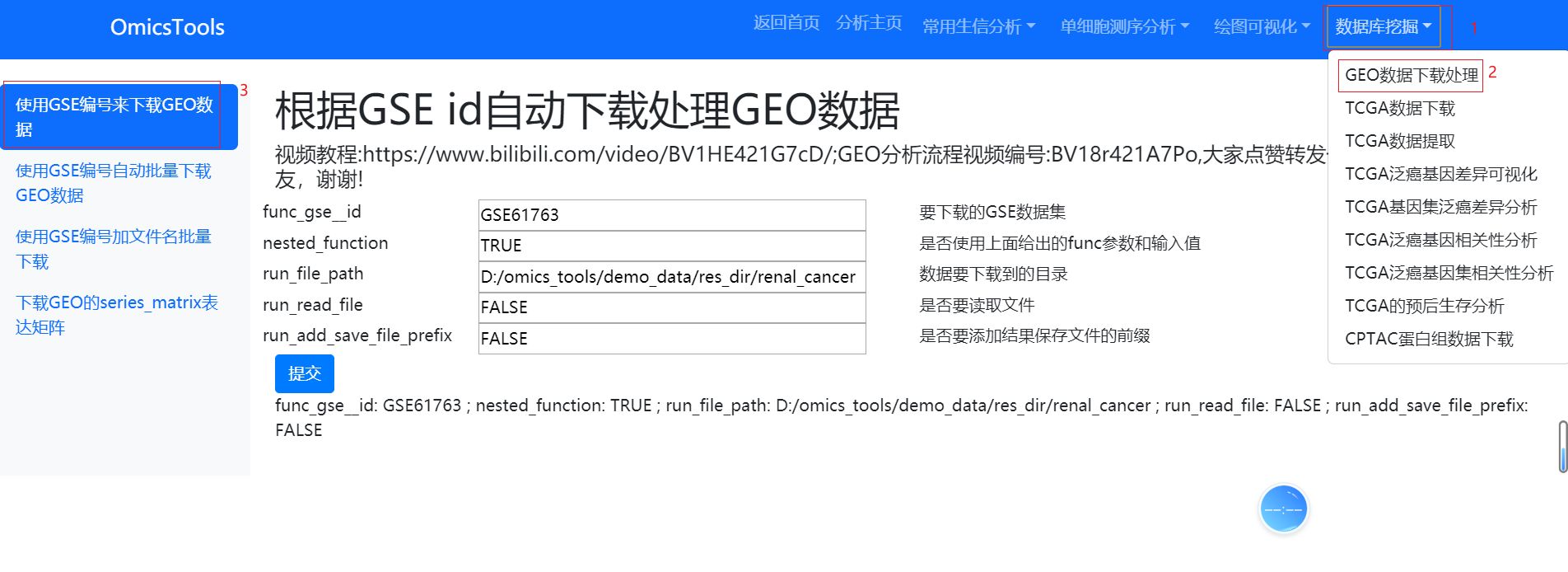
该模块的教程
- 知乎地址:根据GEO的GSE数据集编号自动下载和处理GEO数据教程: https://zhuanlan.zhihu.com/p/708053447
该根据GSE id自动下载处理GEO数据的GEO下载模块的常见问题答疑详解
GEO的数据集格式多种多样,使用该模块下载GEO数据集最好的情况下得到的结果是什么样的?
最好的结果就是下图这样,直接通过该模块点击下载后,会得到下面这些下载提取整理好的文件:
- 一次性得到注释好基因名称的表达矩阵文件,如GSE61763_exp_matrix.csv,GSE61763_exp_matrix.rds
- 得到样本的注释信息文件: 如GSE61763_sample_info.csv,
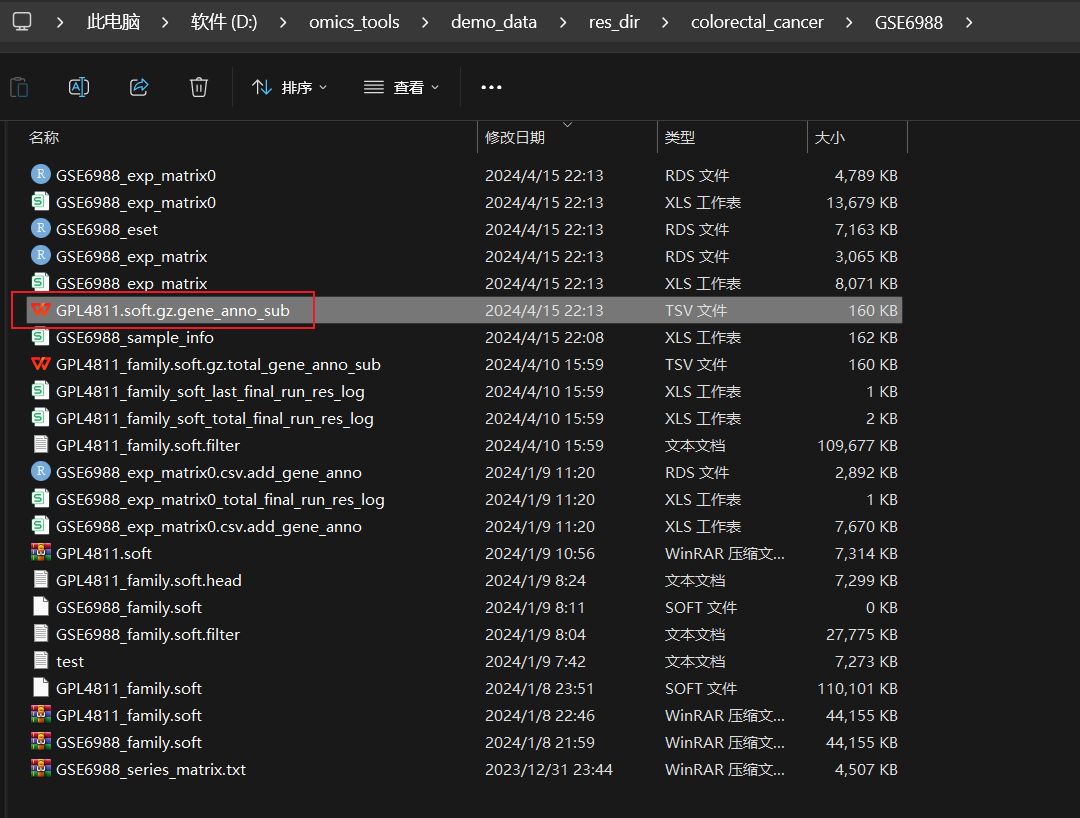
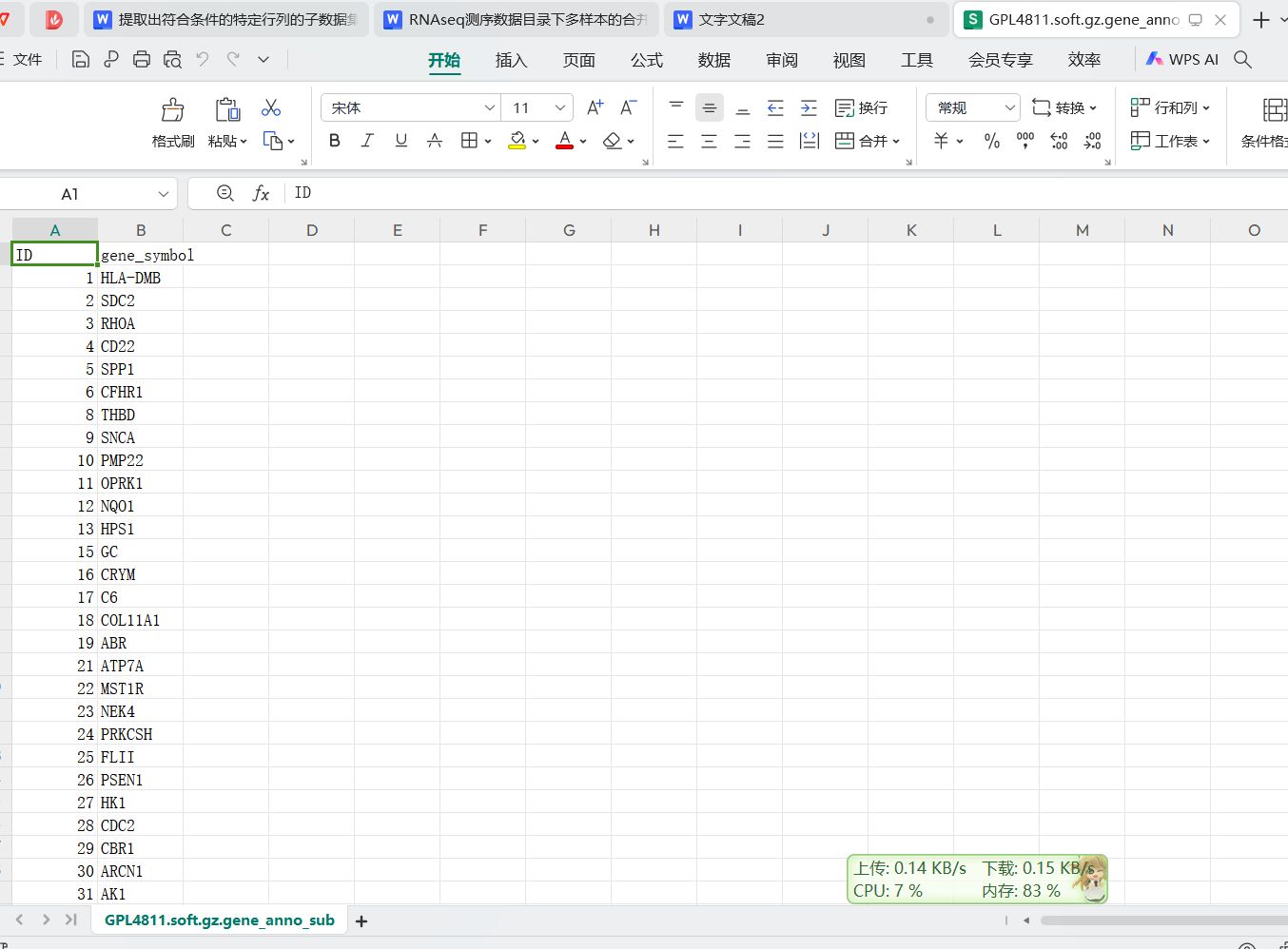
- 得到从GPL soft注释文件中提取到的基因探针跟基因名称的对应关系的两列数据文件,如GPL191197.soft.gz.gene_anno_sub.tsv文件
- 得到了使用表达矩阵,基因注释信息,样本注释信息等数据构建的ExpressionSet对象格式文件,如GSE61763_eset.rds 文件
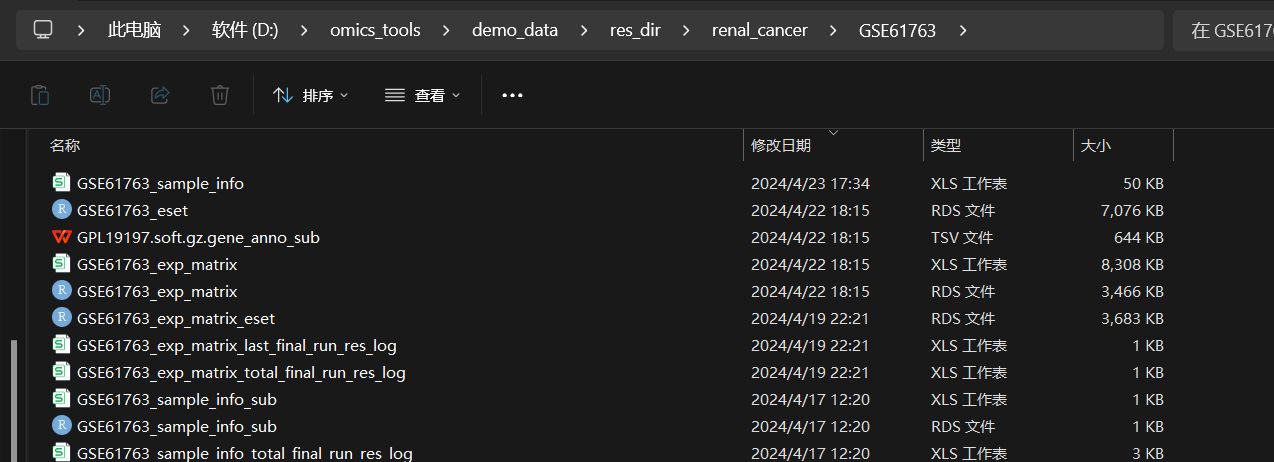
结果文件列表
GEO的数据集格式多种多样,使用该模块下载GEO数据集第二好的情况下得到的结果是什么样的?遇到这种情况该怎么处理?
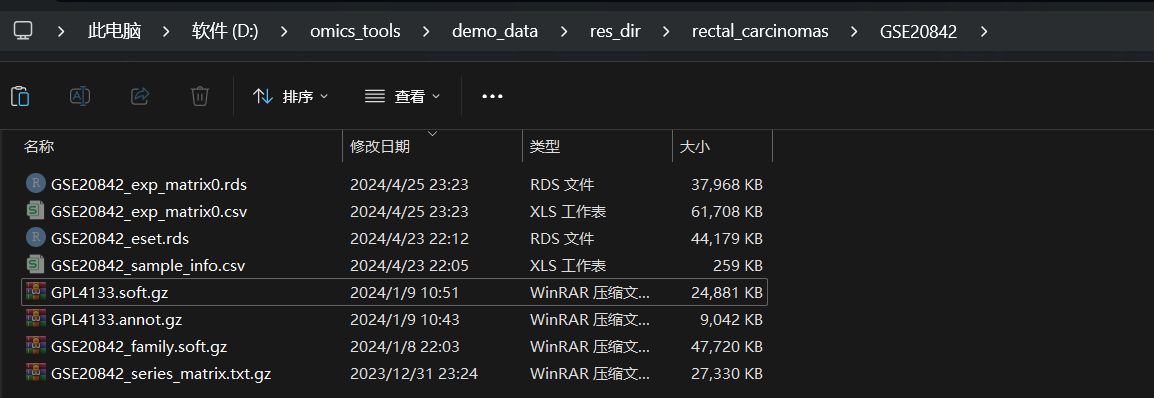
- 提取到了GEO数据集的表达矩阵文件,如GSE20842_exp_matrix0.csv,GSE20842_exp_matrix0.rds
- 得到样本的注释信息文件: 如GSE20842_sample_info.csv,
- 得到了使用表达矩阵,探针信息,样本注释信息等数据构建的ExpressionSet对象格式文件,如GSE20842_eset.rds文件
但是这个表达矩阵有个问题就是,行名不是基因名称,而是探针名称,所以我对这个文件名称做了一些修饰,如果文件名后缀是exp_matrix0.csv,那么这样的表达矩阵里面基本上是没有注释好基因名称,需要在手动下载一下GEO的GPL文件从GPL文件中提取出基因的注释信息再跟表达矩阵合并,这样的处理作为我也有详细的处理教程来教大家怎么做

如果文件后缀就是exp_matrix.csv,那么就是已经成功把基因名称提取出来并整合到表达矩阵中了。对于没有基因名称只有基因探针的表达矩阵,可以用下面这两个模块进行处理:
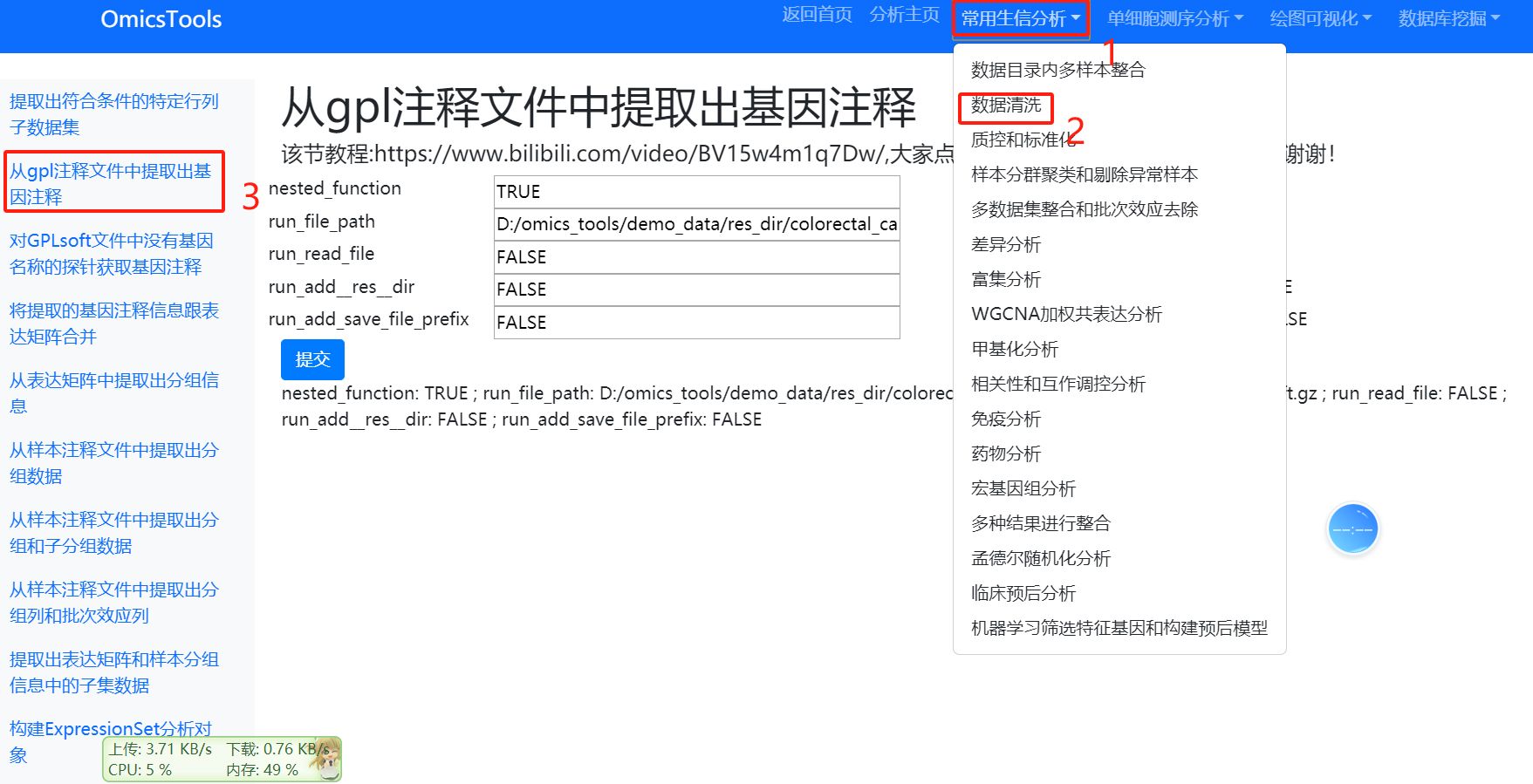

基因探针soft.gz注释文件下载
GPL背景知识介绍
在基因表达数据分析中,了解特定的基因探针平台(GPL)的注释信息是非常重要的。`GEO`,或者称为`Gene Expression Omnibus`,是一个存储高通量基因表达数据(如微阵列和次世代测序数据)的公共数据库,由美国国立生物技术信息中心(NCBI)管理。每个数据集通常与一个或多个探针平台相关联,这些平台在GEO中被指定为GPL(Gene Expression Omnibus Platform)。
GEO的GPL(Gene Expression Omnibus Platform)
在GEO中,每个基因探针平台被赋予一个唯一的GPL编号。GPL描述了用于测量基因表达的技术和方法,包括探针的设计、制造信息以及探针与基因(或转录本)之间的对应关系。这些信息对于正确解释基因表达数据至关重要。
主要内容包括:
1. **平台描述**:
- **制造商**:如Affymetrix, Illumina, Agilent等。
- **技术类型**:如微阵列芯片、次世代测序。
- **探针特征**:探针数量、探针覆盖的生物体(如人类、小鼠等)。
2. **探针注释**:
- **探针ID**:每个探针在平台上的唯一标识。
- **探针位置信息**:探针在基因组或转录组上的具体位置。
- **靶标基因**:每个探针旨在检测的基因或转录本。
- **相关序列**:探针的核苷酸序列。
3. **访问和使用**:
- **GPL访问编号**:如GPL570。
- **数据集示例**:与该平台相关的GEO数据集编号(如GSE号)。
- **下载和查询**:用户可以通过NCBI的GEO查询界面查询特定平台的注释数据。
为什么探针注释重要?
- **精确的生物学解释**:正确的探针到基因的映射确保了实验结果的生物学解释的准确性。
- **数据比较**:统一的探针注释允许不同实验数据间的有效比较。
- **研究复现**:详细的平台注释信息帮助其他研究者复现和验证实验结果。
理解GEO的GPL信息有助于研究人员有效利用公共基因表达数据
基因探针soft.gz注释文件的自动下载
根据GSE id自动下载处理GEO数据(必须要运行的模块)
该模块的运行窗口截图
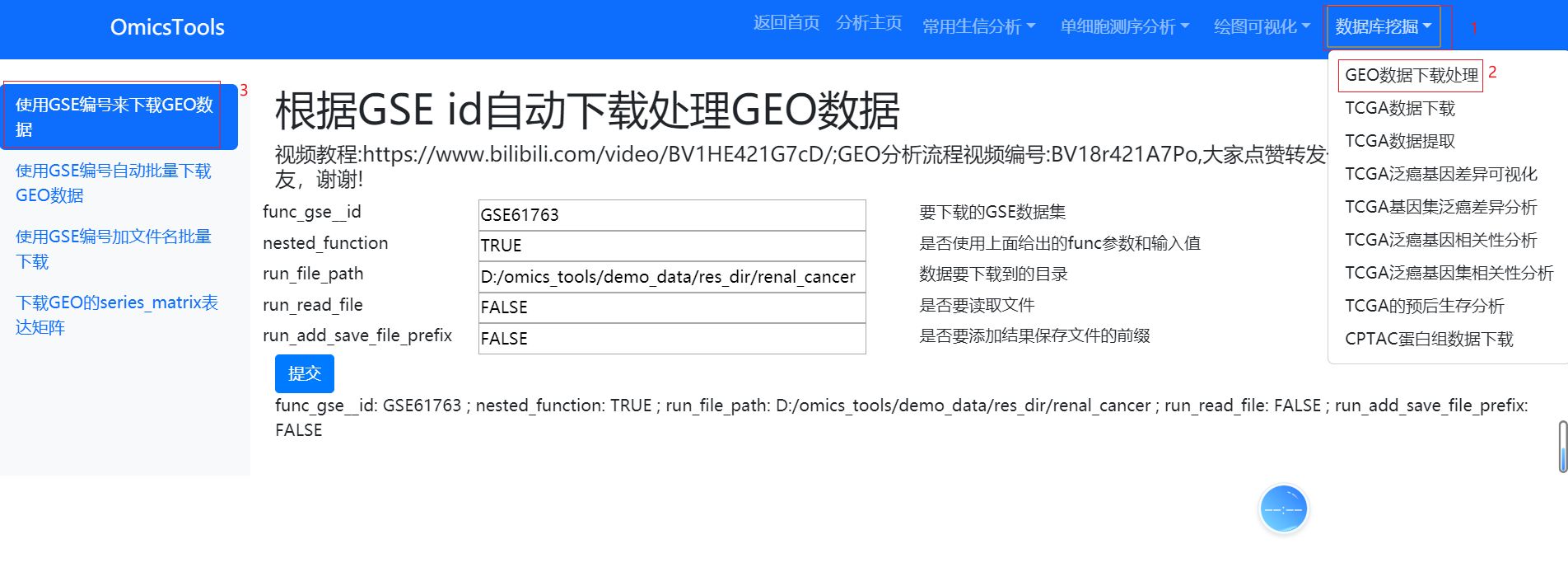
该模块运行的结果文件
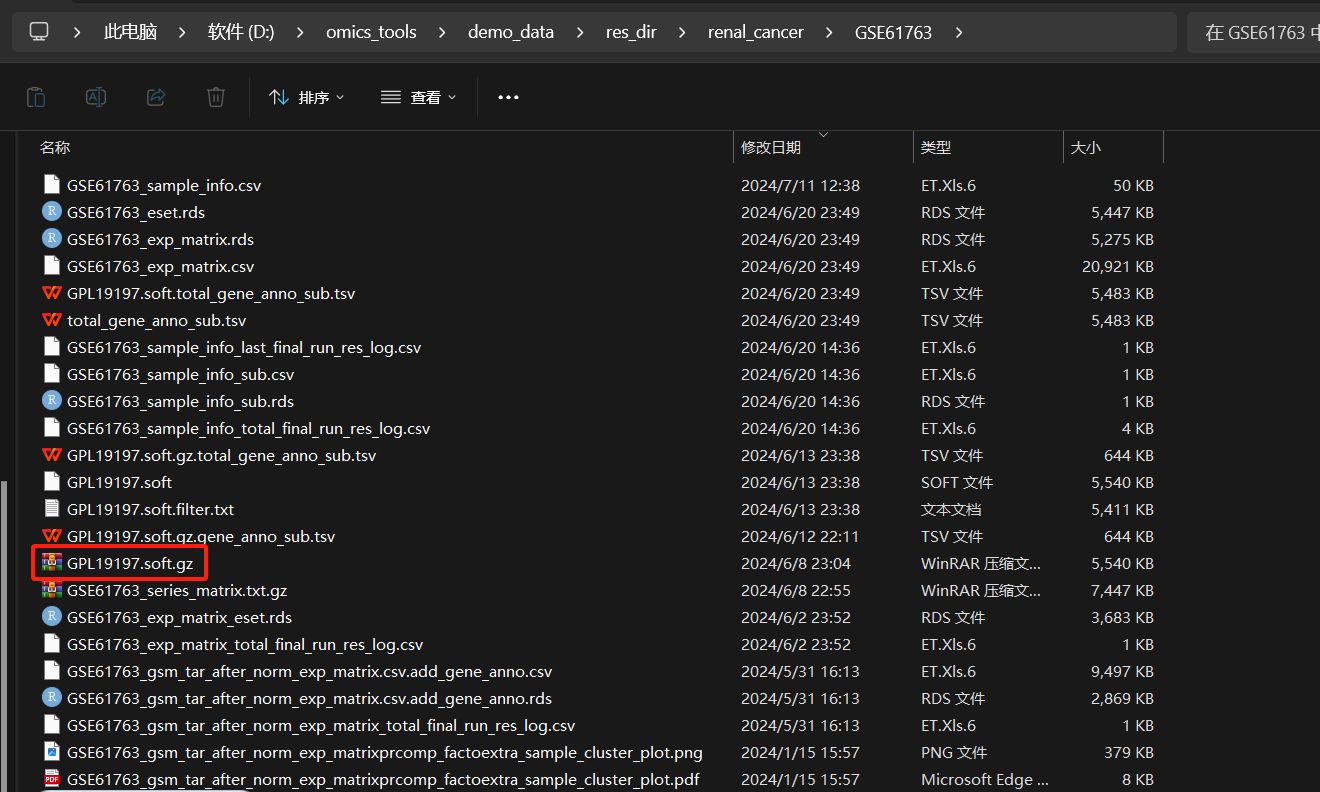
GSE61763数据集的GPL平台编号是GPL19197,在运行该模块执行完成后,会自动下载出GPL191197.soft.gz或GSE61763_family.soft.gz这样的探针注释文件。
GPL探针soft.gz注释文件的手动下载
如果有的时候使用上面的自动下载模块没有下载到GPLXXX.soft.gz或GSEXXX_family.soft.gz注释文件,我们也可以使用手动下载模块来下载GPL的注释文件
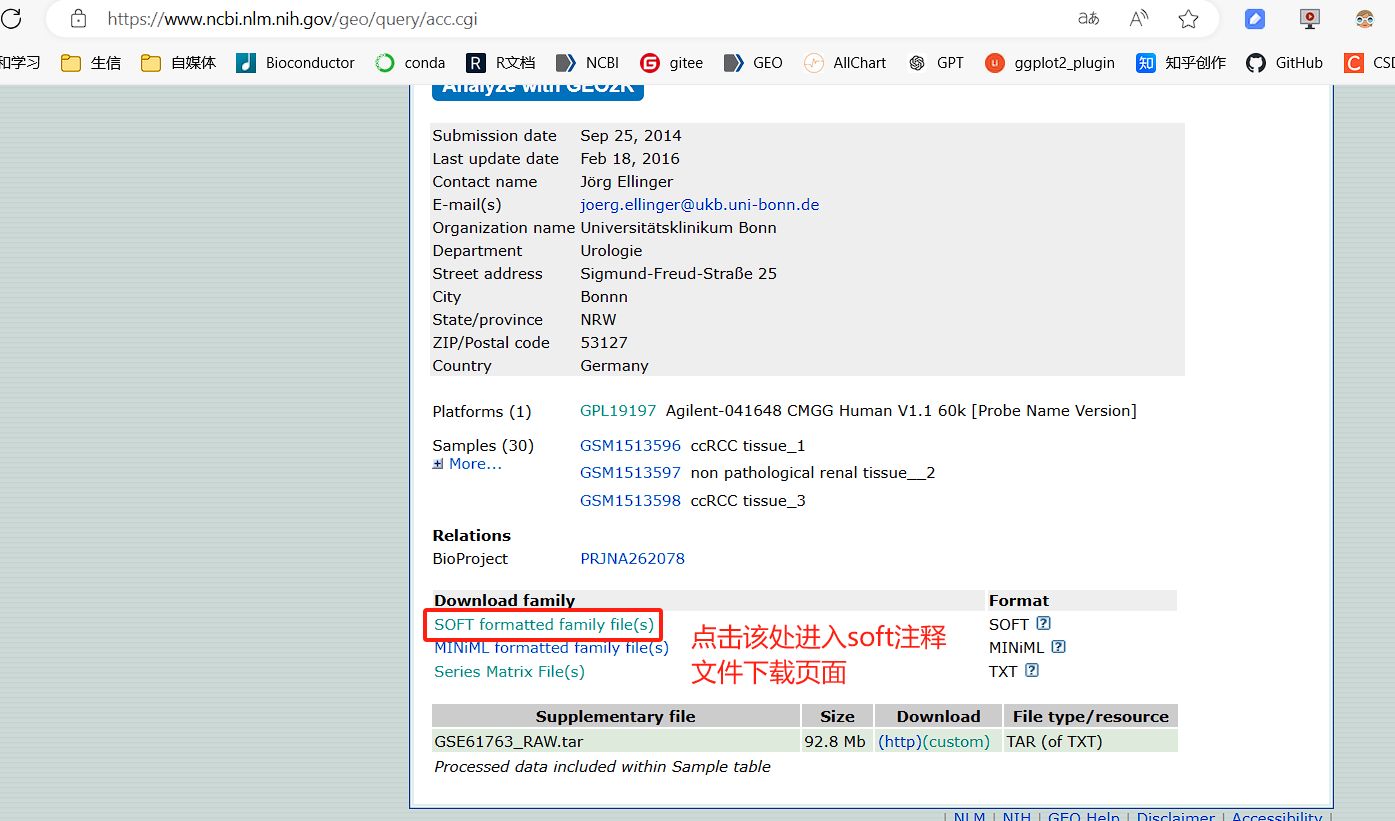
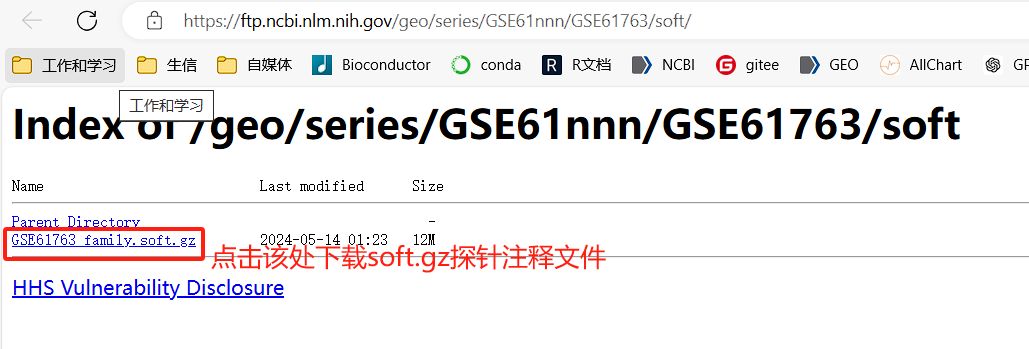
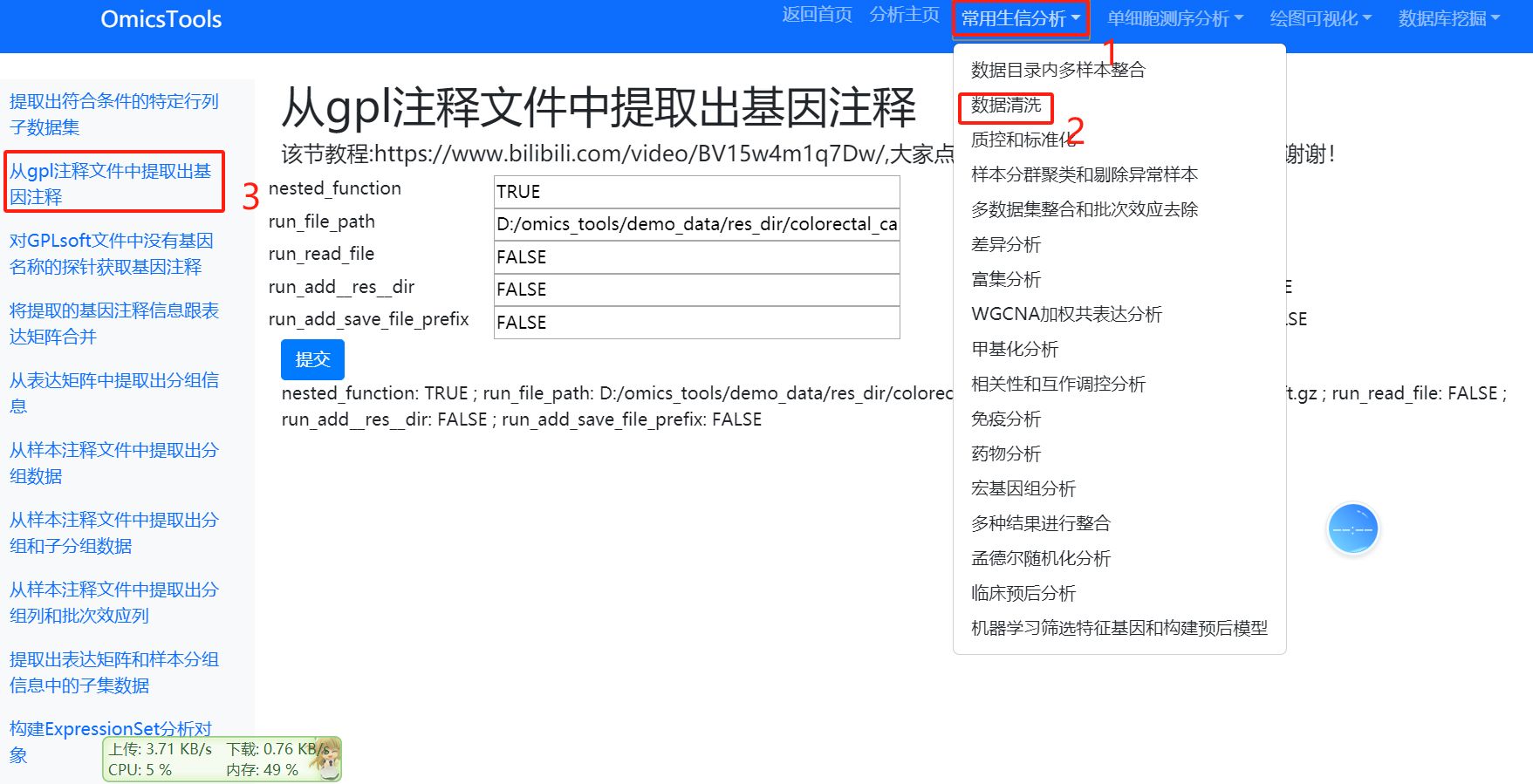
从gpl注释文件中提取出基因注释
教学教程视频
从gpl注释文件中提取出基因注释:https://www.bilibili.com/video/BV15w4m1q7Dw/
运行窗口
运行完成提取到的注释结果
将提取的基因注释信息跟表达矩阵合并
该部分的教学视频
将提取的基因注释信息跟表达矩阵合并:https://www.bilibili.com/video/BV12f421Q7nK/
窗口截图展示
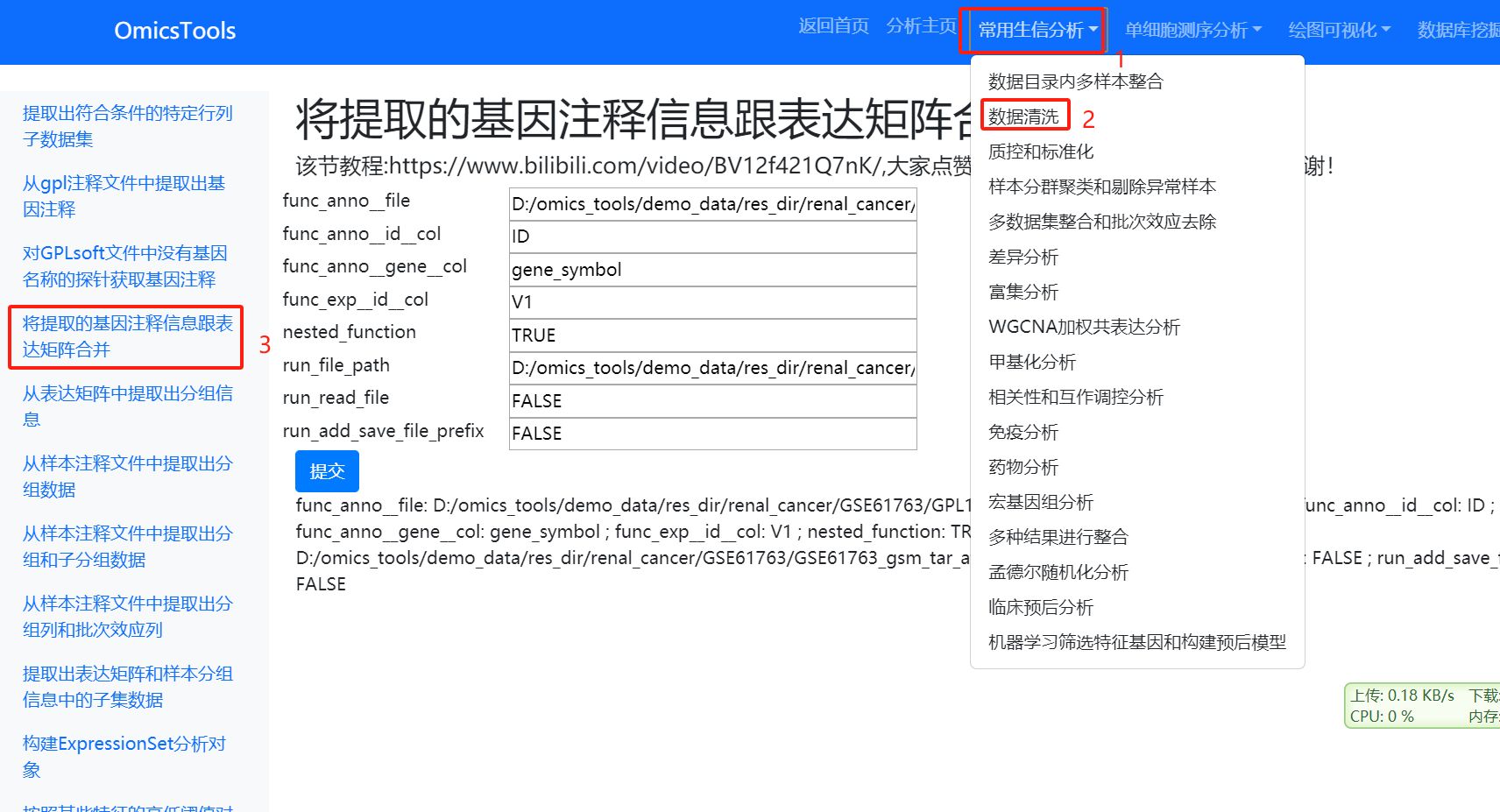
参数注释信息解释
func_anno__file: 提取出的基因注释信息文件
func_anno__id__col:基因注释文件中基因id的列名
func_anno__gene__col:基因注释文件中gene_symbol所在的列名
func_exp__id__col:表达文件中的索引列
nested_function:是否嵌套函数
run_file_path:表达数据文件所在的路径
run_read_file:是否要读取文件
run_add_save_file_prefix:是否要添加结果保存文件的前缀
提交
参数给出的默认值
func_anno__file:D:/omics_tools/demo_data/res_dir/renal_cancer/GSE61763/GPL19197.soft.gz.total_gene_anno_sub.tsv ;
func_anno__id__col: ID ;
func_anno__gene__col: gene_symbol ;
func_exp__id__col: V1 ;
nested_function: TRUE ;
run_file_path: D:/omics_tools/demo_data/res_dir/renal_cancer/GSE61763/GSE61763_gsm_tar_after_norm_exp_matrix.csv ;
run_read_file: FALSE ;
run_add_save_file_prefix: FALSE
运行中的信息展示
分析正在执行中,请稍后, 运行结果保存的目录位置为: D:/omics_tools/demo_data/res_dir/renal_cancer/GSE61763\res_dir; 运行结果日志保存的路径为: D:/omics_tools/demo_data/res_dir/renal_cancer/GSE61763\res_dir\GSE61763_gsm_tar_after_norm_exp_matrix_last_final_run_res_log.csv
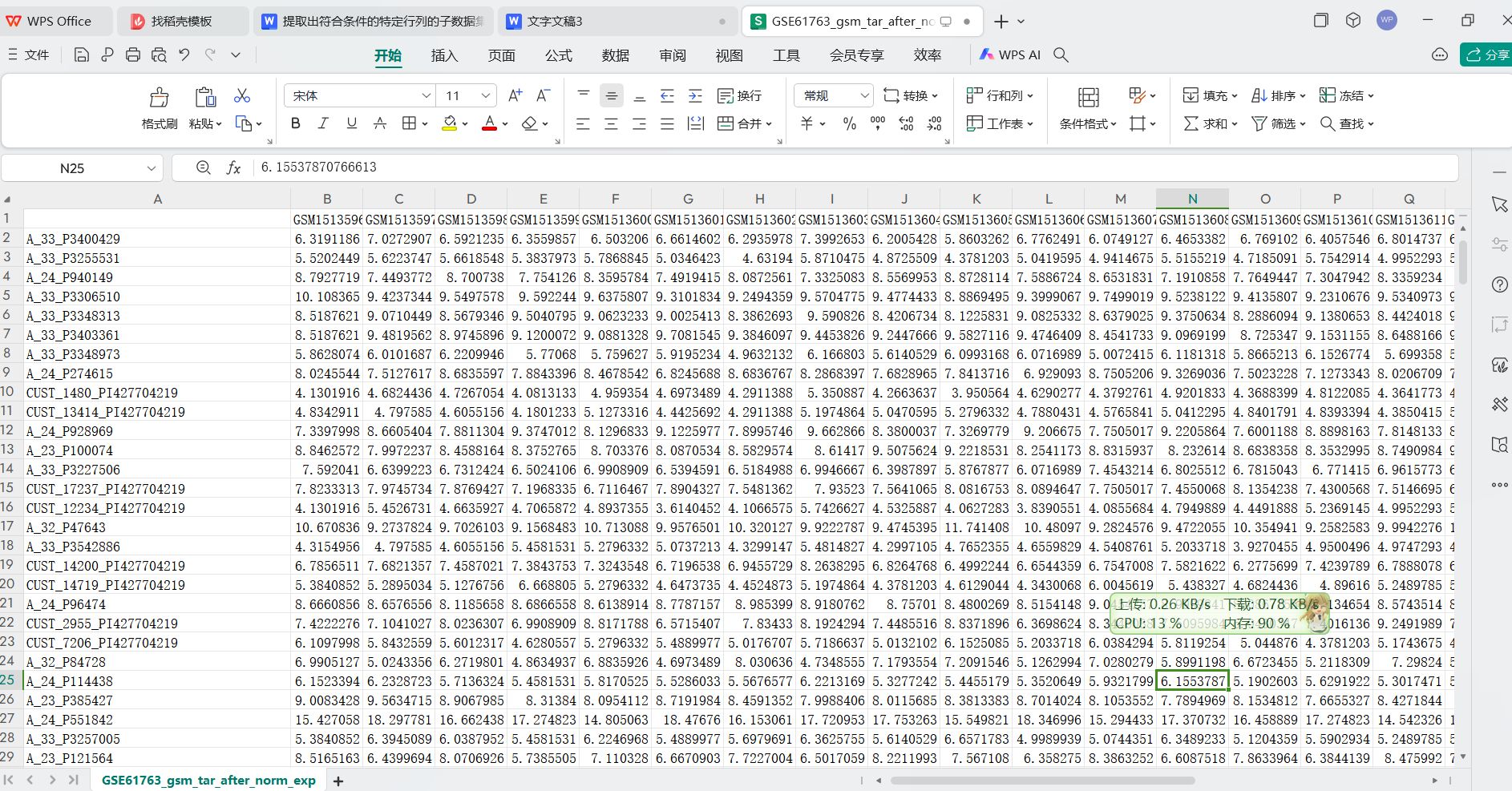
表达数据集的内容展示
D:/omics_tools/demo_data/res_dir/renal_cancer/GSE61763/GSE61763_gsm_tar_after_norm_exp_matrix.csv
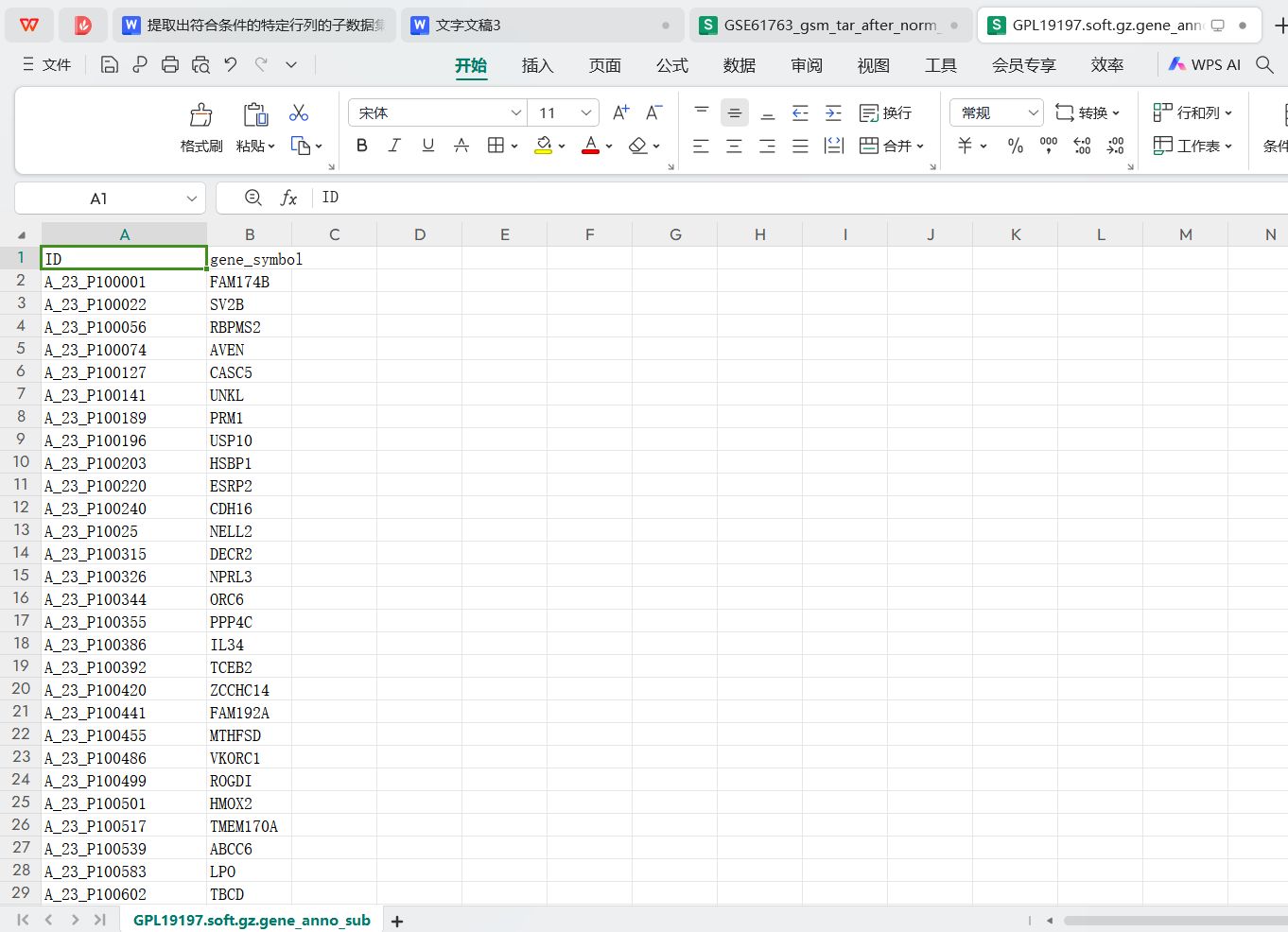
基因注释文件信息
运行完成的信息显示
执行已完成,运行结果保存的目录位置为: D:/omics_tools/demo_data/res_dir/renal_cancer/GSE61763\res_dir; 分析结果日志保存的路径为: D:/omics_tools/demo_data/res_dir/renal_cancer/GSE61763\res_dir\GSE61763_gsm_tar_after_norm_exp_matrix_last_final_run_res_log.csv
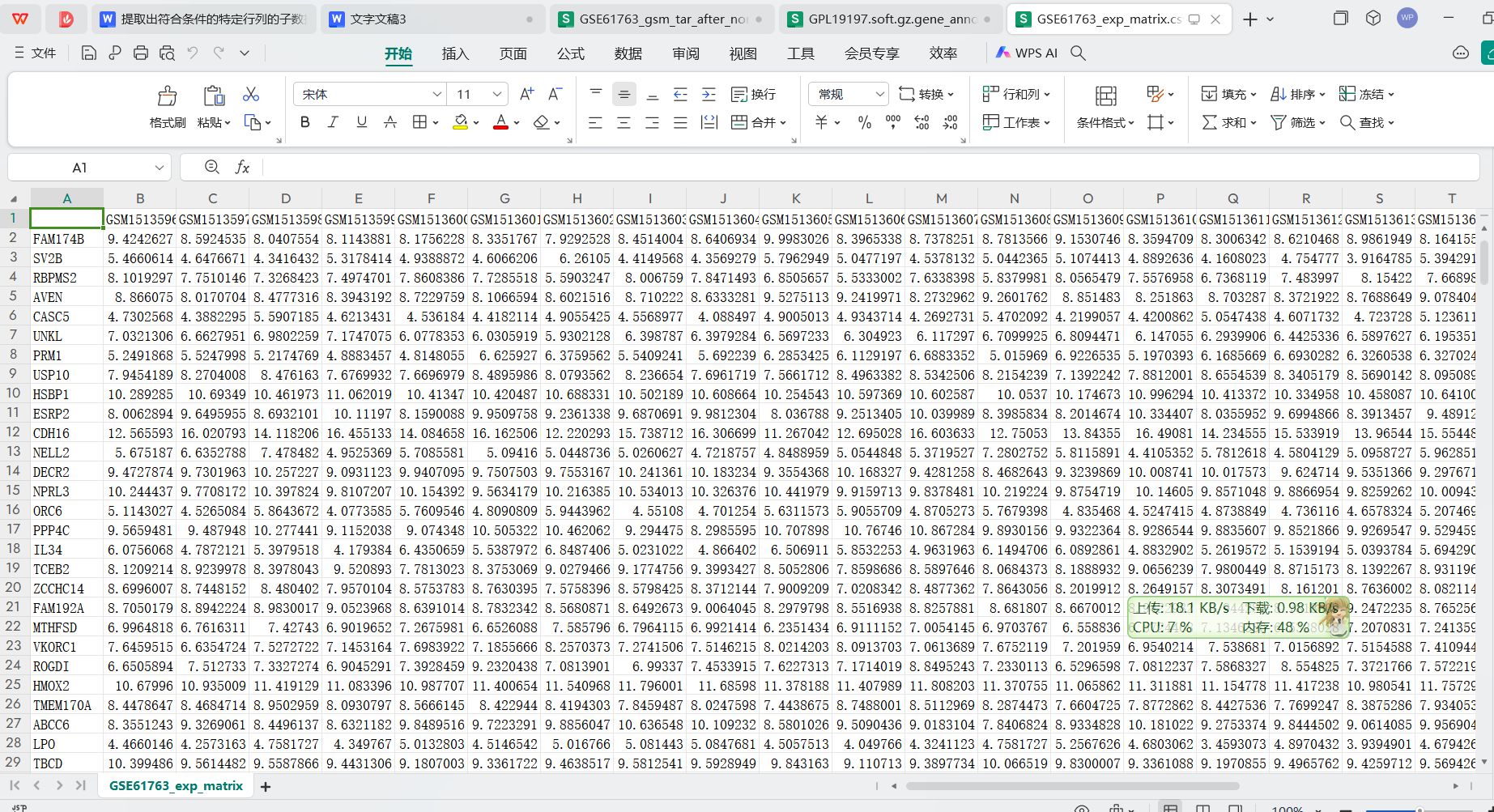
运行完成的结果展示


















 2万+
2万+

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











