







scJoint integrates atlas-scale single-cell RNA-seq and ATAC-seq data with transfer learning
Nature Biotechnology
2022/01/20
https://www.nature.com/articles/s41587-021-01161-6#data-availability
scJoint:基于迁移学习的标签转移模型。
Motivation
- it has been noted that the extreme sparsity of scATAC-seq data often limits its power in cell-type identification // scATAC 数据存在“极端稀疏性”从而导致无法实现细胞分群
- However, direct applications of these methods to multiomics data integration are computationally challenging and often suboptimal, since different modalities have vastly different dimensions and sparsity levels. // 因为不同的测序方法产生不同的数据维度或者稀疏性的差异,因此基于批次矫正或单细胞多组学整合的方法并不是最优的方法(批次矫正或者整合的方法必然带来信息的损失)
Innovations
- 用于降维的 loss function
- 相似性 loss function,在细胞没有 overlap 时提供更加灵活的 label 对齐
Main step of scJoint
scJoint requires simple data preprocessing, with the input dimension equal to the number of genes in the given datasets after appropriate filtering. Chromatin accessibility in scATAC-seq data is first converted to gene activity score // 模型的输入数据是单细胞表达矩阵。在迁移学习时,ATAC矩阵要转换为基因活性得分
-
Step 1 performs joint dimension reduction and modality alignment in a common embedding space through a new neural-network-based dimension reduction (NNDR) loss and a cosine similarity loss respectively. // 通过降维神经网络损失函数和 cosine 相似性损失函数在传统的 embedding 空间进行数据对齐。

降维神经网络损失(NNDR)与 PCA 相似,是通过提取含有最大方差的正交特征。
其中红色框框用于计算最大的组间方差:
第一个框计算每一个细胞与几何中心的距离
第二个框计算两个不同细胞 embedding 之间的相似度
其中绿色框框用于计算最小重构代价,用于稳定样本空间
这一步仅有 scRNA 数据参与训练
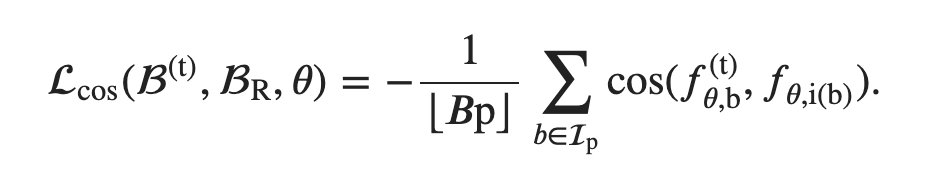
将 ATAC 数据进行嵌入,使用余弦相关性计算嵌入后的 ATAC 数据与 scRNA 数据的相关性。这一步是为了保证嵌入空间最大程度上能够使得 ATAC 数据与 scRNA 数据相关,所以这里取
负值最终嵌入模型的损失函数如下:
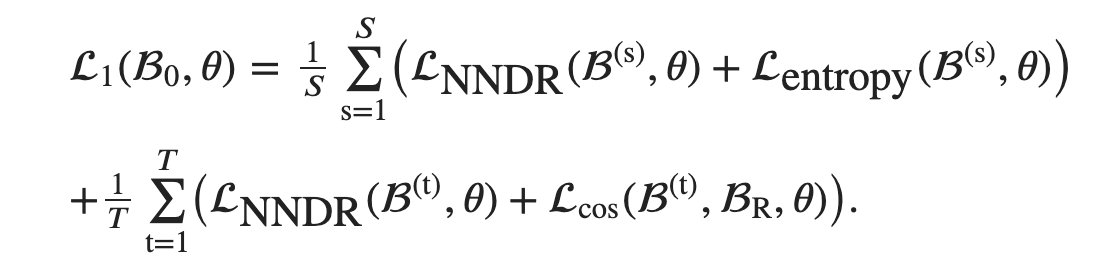
算式中分为 2 个部分:
- 计算 scRNA-seq 的嵌入损失函数与分类交叉熵,在确保分类准确的同时稳定嵌入空间
- 第二部分计算 ATAC 的嵌入损失函数,以及ATAC 数据与所有的 scRNA 数据的余弦相似性损失函数
-
In Step 2, treating each cell in scATAC-seq data as a query, we identify the k-nearest neighbors (KNN) among scRNA-seq cells by measuring their distances in the common embedding space, and transfer the cell-type labels from scRNA-seq to scATAC-seq via majority vote. // 将所有的 scATAC-seq 数据当做“待调查”样本,通过 KNN 算法测量每一个样本与 scRNA-seq 的距离,通过多数投票的方法给“待调查”样本,从而确定每一个样本的“伪标签”。
-
In Step 3, we further improve the mixing between the two modalities by using the transferred labels in a metric learning loss. // 修改损失函数后进一步的优化对齐空间
在第二步中,配对好伪标签后重新训练嵌入空间,此时将所有的数据全部纳入计算
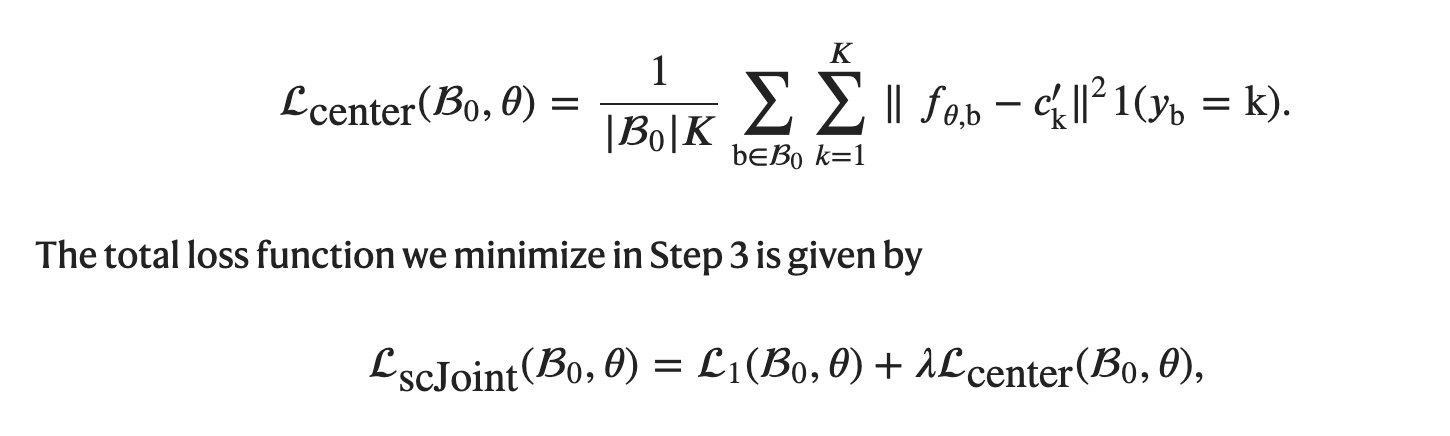
上面的算式计算了各个嵌入向量与其几何中心的距离
下面的算式与第一步相似,此时计算交叉熵时同时计算 ATAC 的交叉熵。
Compared scJoint with other methods
文章主要通过与常用的 3 种数据整合方法进行对比:
整合两套独立的老鼠单细胞图谱数据:
- scRNA-seq :96404 个细胞,20 种不同的器官,73 种细胞类型,共使用 2 套不同的流程获得的数据(具有较强的批次效应)
- scATAC-seq:81173 个细胞,13 种不同的组织,29 种细胞类型
所使用的的数据集中,包括 19 种共同的细胞类型( 101,692 cells)。
模型评估-1
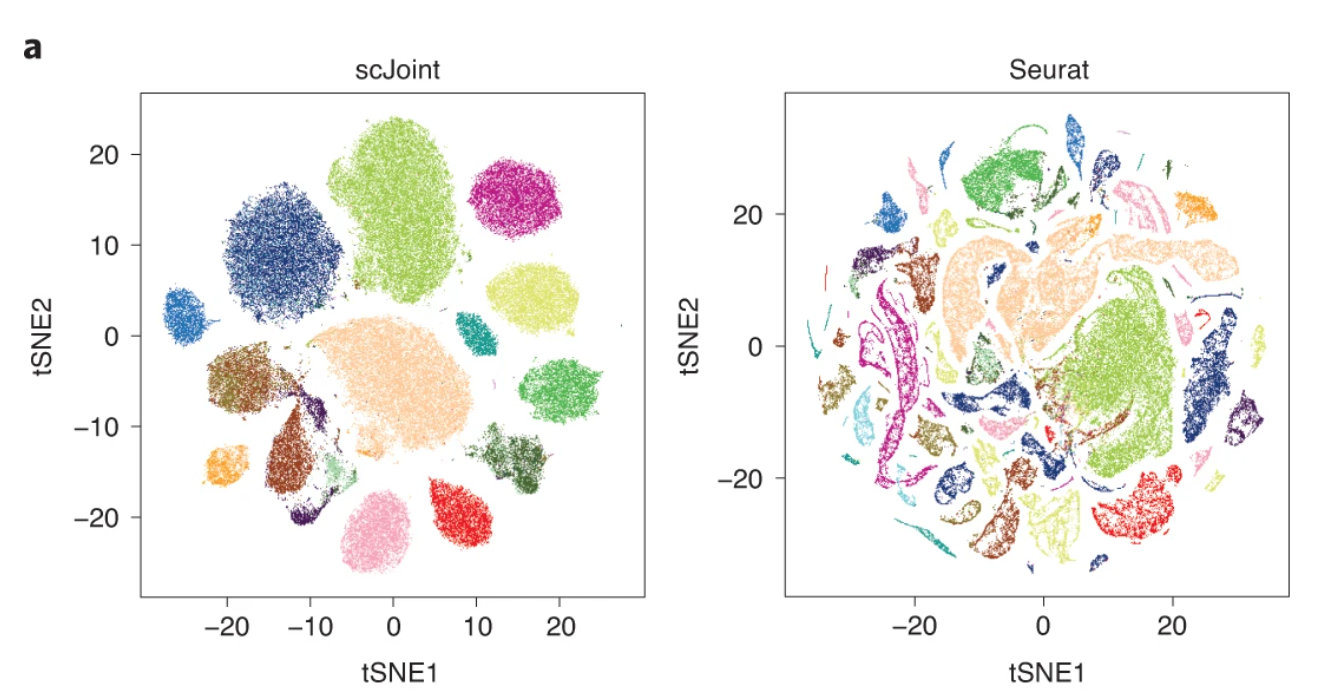
文章首先评估了共有的 19 种细胞类型。使用 scJoint 将scATAC-seq 数据与 scRNA-seq 的标签对齐。
显然,相比于另外三种整合方法,scJoint 的对齐方法效果明显更好
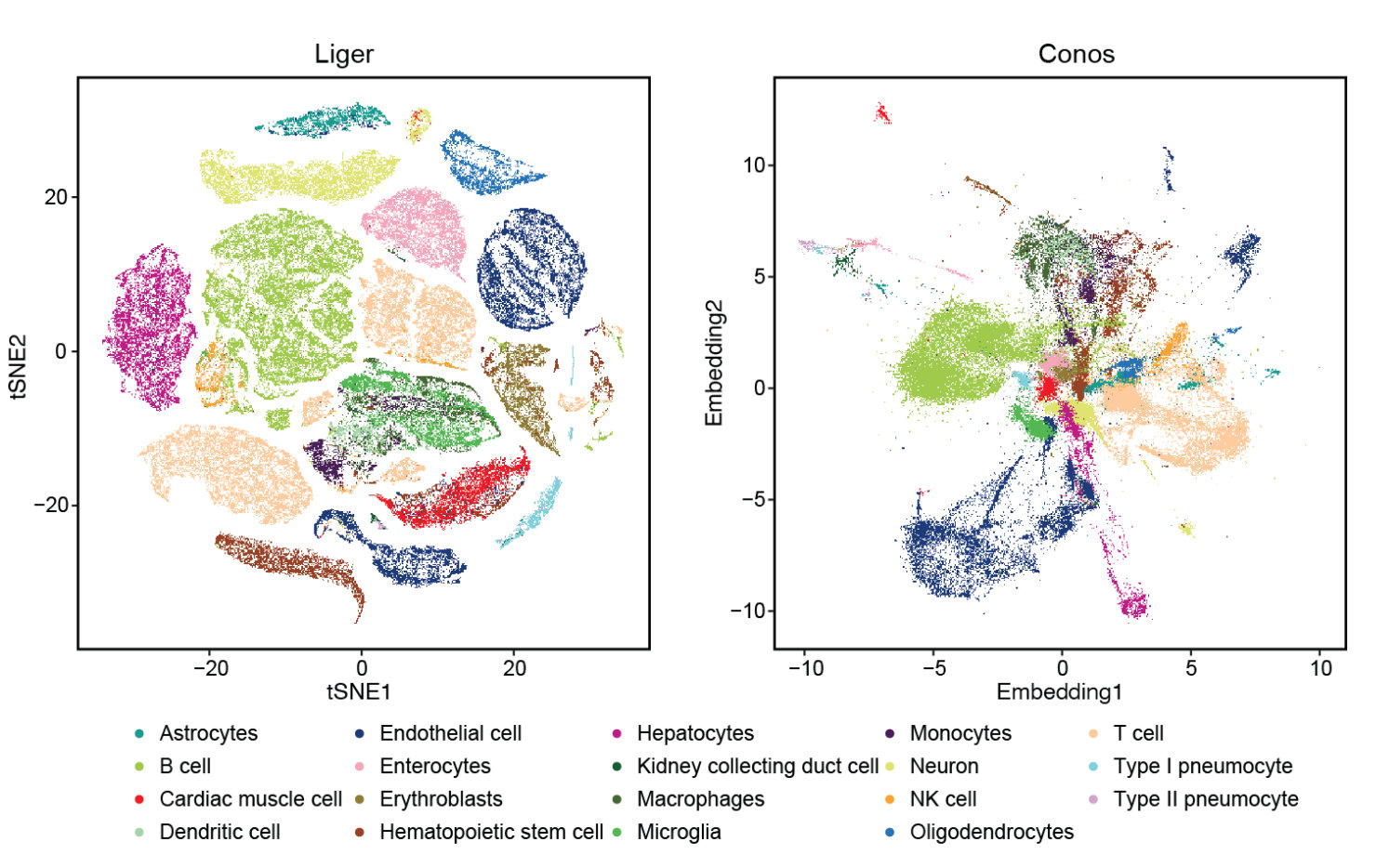
文章也通过计算轮廓相似度和 F1 值(与文章 label 比较)进行数值化评估
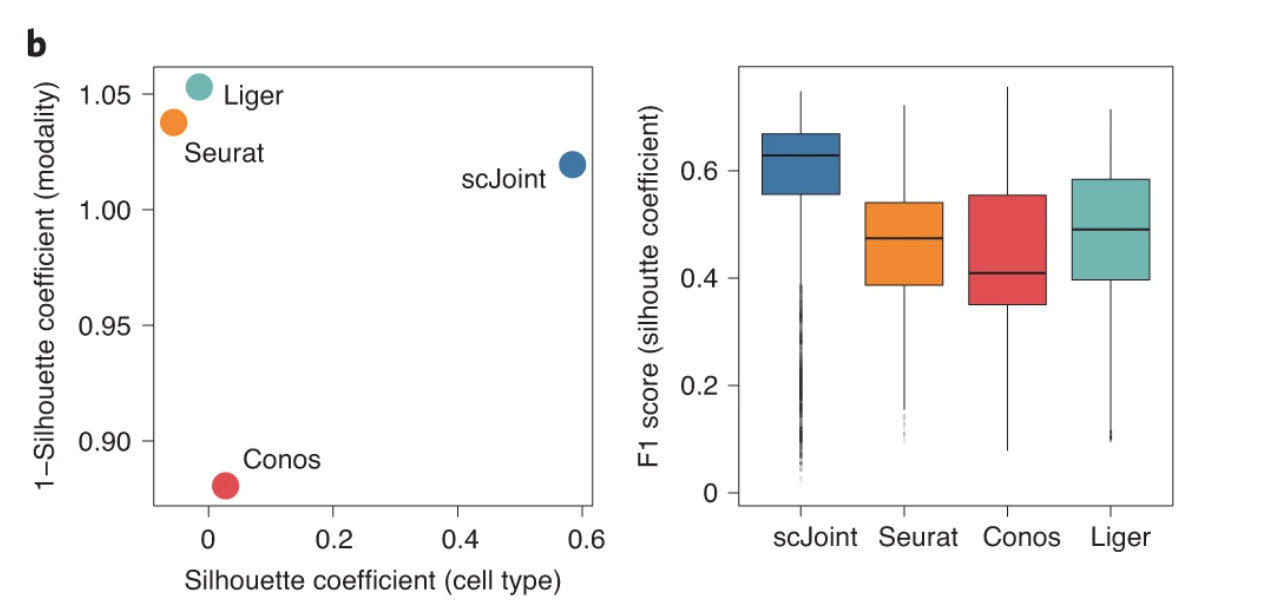
图 B 左:横坐标代表的是细胞类型间的轮廓系数[-1,1],纵坐标代表的是 1-不同测序方法之间的轮廓系数(没啥意义)。总的来看,可以看出在细胞类型间,scJoint 的轮廓系数最大(类间距离大,类内距离小)。不同测序方法的离散程度几乎相同(根据公式不难算出,测序方法的轮廓系数接近0)。
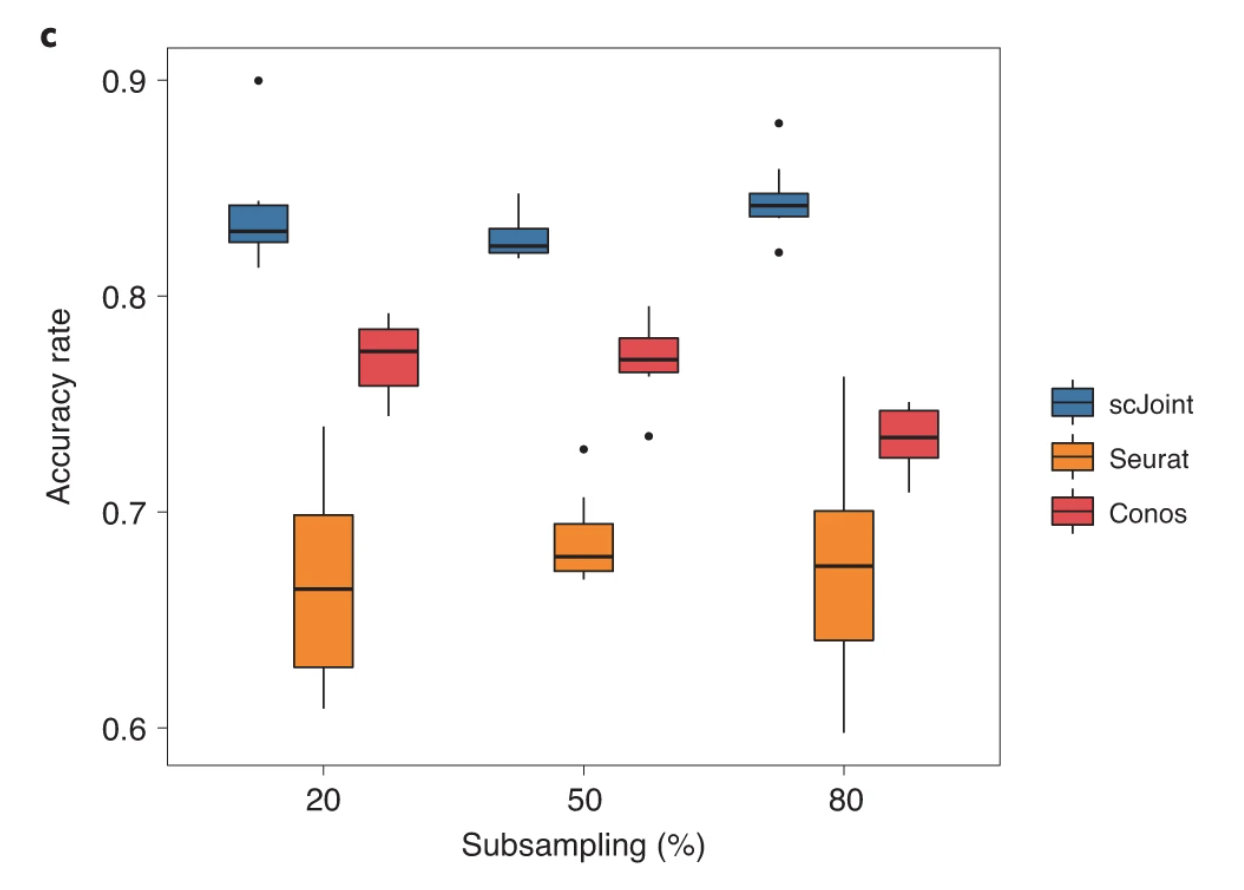
在不同的样本容积下,与文章的label 相比三种数据整合的方法相对稳定 scJoint 的分类准确率最高。再给予错误标签的情况下 scJoint 仍可以维持较高的稳定性(Supplementary Figure S4)
奇怪的是为什么没有比较 Liger
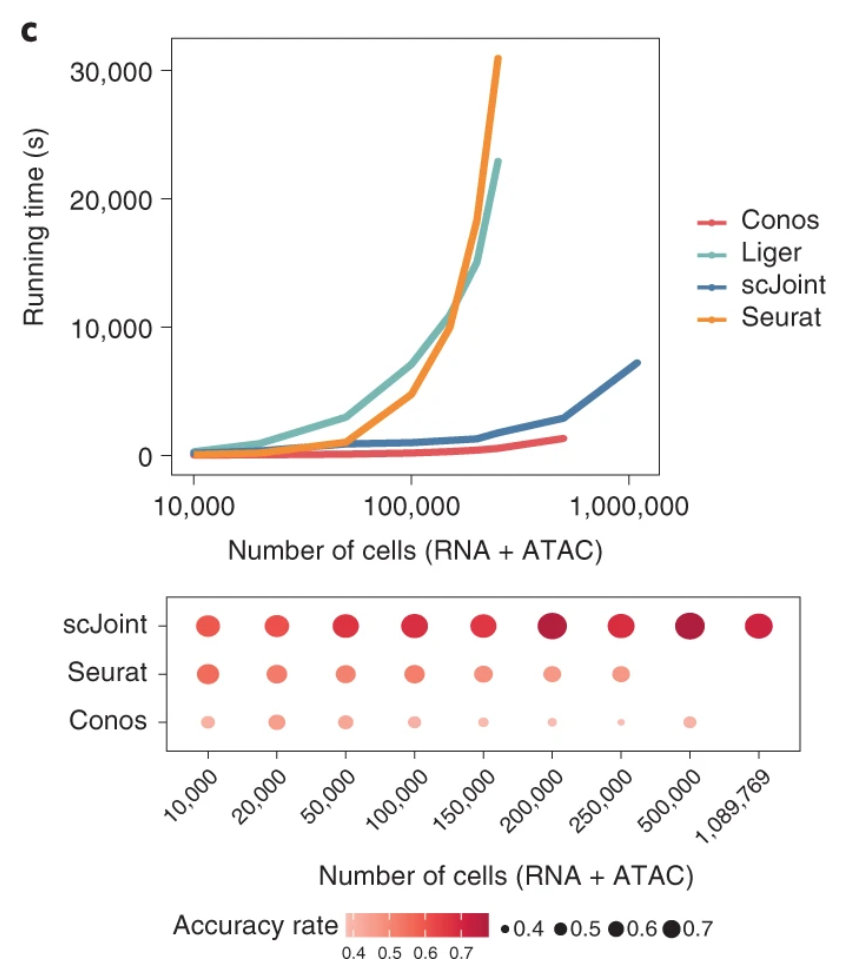
另外,scJoint 的运行速度并不会随着数据量的增加呈指数级增加。因此,scJoint 更加的适合百万级别的数据
模型评估-2
上文提到,在单细胞数据中,有多达 79 种细胞类型,然而在 ATAC 数据中仅有 29 种细胞类型,并且数据中还存在部分‘Unknow’的细胞。因此作者进行了全数据评估。
直白一点的意思就是,把所有的 ATAC 数据的 label 全部当‘Unknow’仅仅根据 scRNA-seq 数据进行训练,然后将 ATAC 的数据与 scRNA-seq 数据对齐,从而获取各个 ATAC 的 label
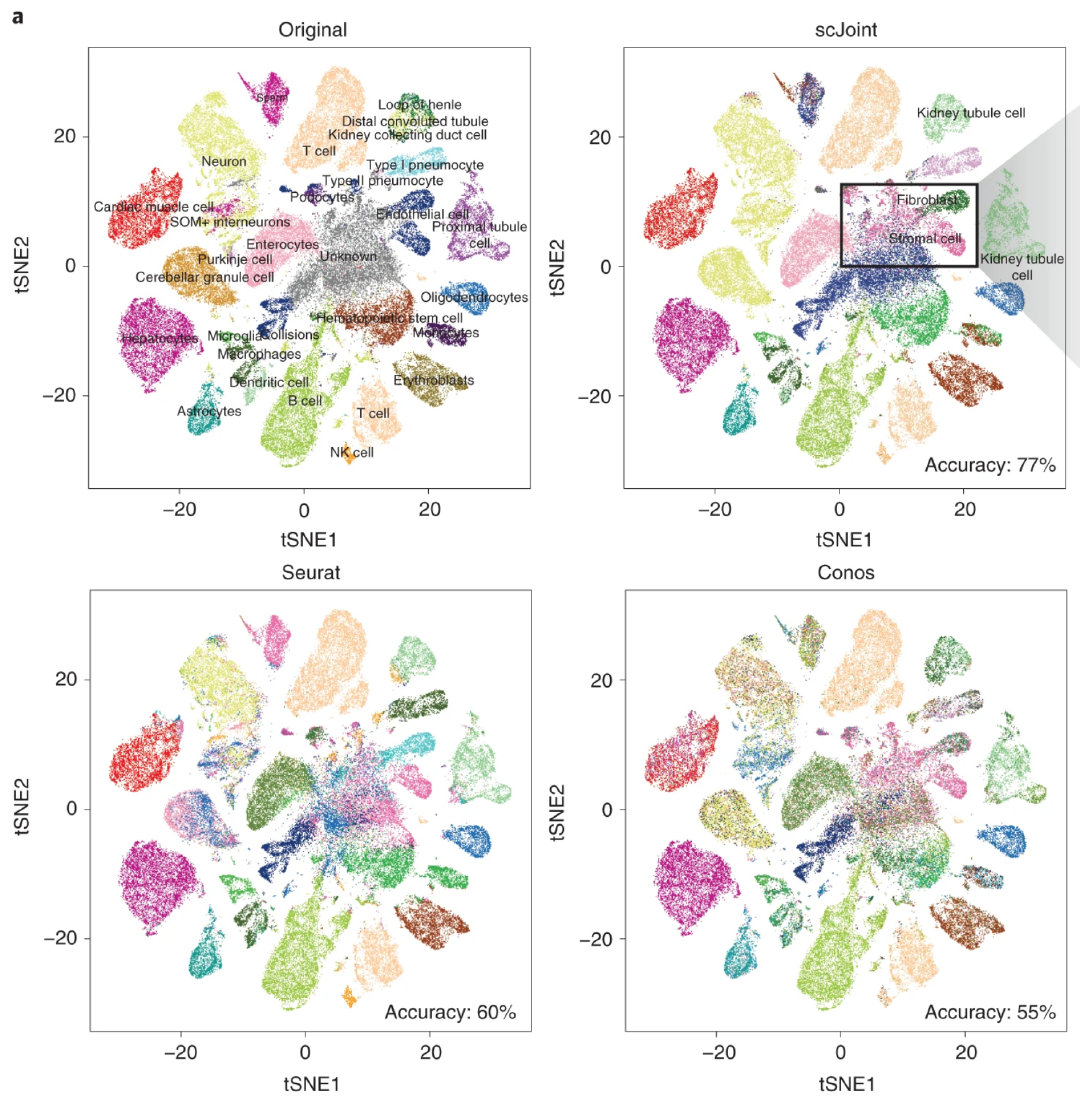
同样的,看图的话 scJoint 分群还是比较明显的,一部分原始标签为“Unknow”的细胞scJoint也进行了分群,并在基因表达层面进行了鉴定。
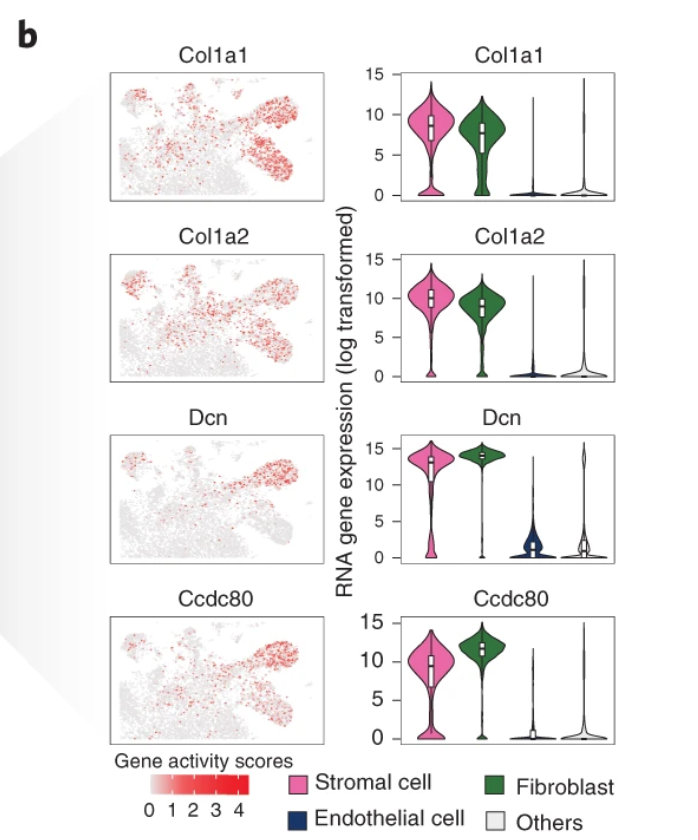
These cells show high gene activity scores for Col1a1, Col1a2, Dcn and Ccdc80, all of which are markers with high expression levels in stromal cells and fibroblasts, but low expression levels in endothelial cells from the scRNA-seq data // 这些细胞显示了Col1a1、Col1a2、Dcn和Ccdc80的高基因活性得分,所有这些都是在基质细胞和成纤维细胞中高表达,在内皮细胞中低表达
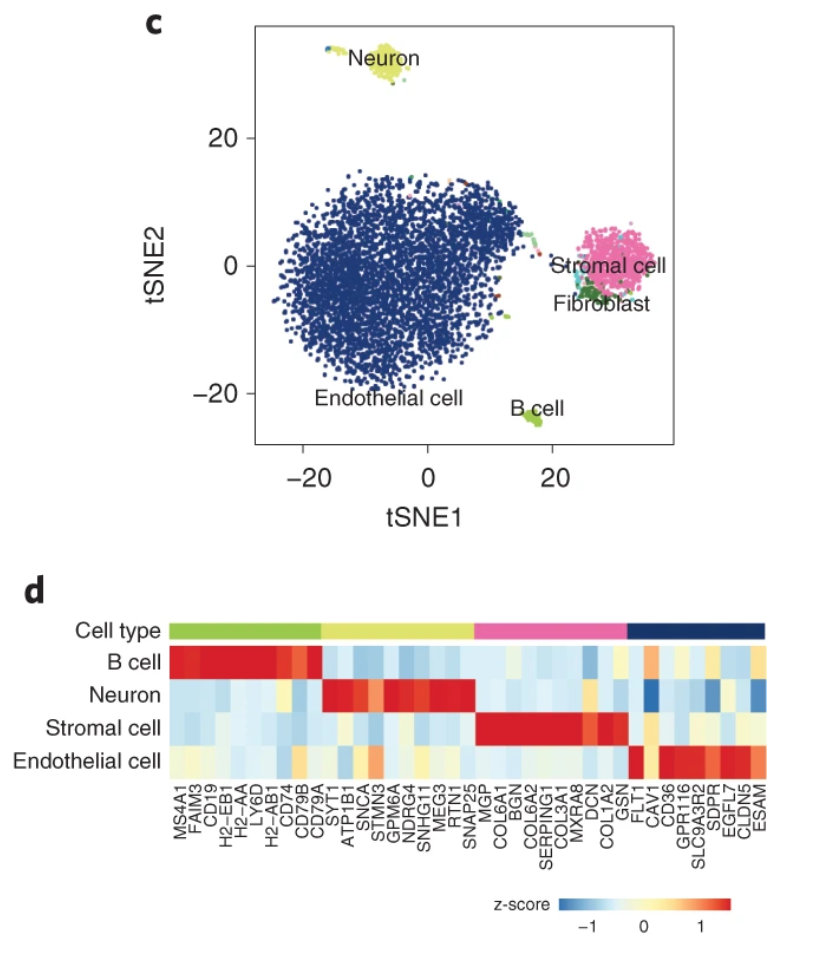
单独的将 “Unknow” 细胞进行 tsne 分群,发现具有较明显的分群情况,且基因表型也证实了 scJoint 的注释结果
总结一下:
- 作者首先在标准数据中对 scJoint 方法进行评估,其准确率可达到84% 且对数据的标签分布具有较强的抗扰动性
- 总体水平的评估也进一步的展示出scJoint 可以在一定程度上在强异质性数据上表现出很强的鲁棒性,且能够在更高的分辨率水平上对scATAC-seq细胞进行注释分群
- 结论:scJoint 可以很好的实现基于 scRNA-seq 数据对 scATAC 数据进行迁移注释
Integration of multimodal data across biological conditions
不同处理条件下,细胞状态会发生变化。
在数据分析的时候,该现象会体现在数据的极端异质性。譬如,相同种类的细胞会以样本形式进行聚集,而非细胞类型。那么这时候就需要进行批次处理或者数据整合。然而,这种整合需要解决至少 2 种主要的潜在问题:
- 如何有效的进行数据整合去除批次,并在最大程度上保留处理差异
- 如何在最大程度上保留各个细胞类型的特异性差异
这两个条件是有点矛盾的,因此理论上是很难完成完美的整合。
对于 scJoint 来说,更倾向解决第二个问题,但是确实在一定程度上解决了第一个问题。作者收集了 CITE-seq和 ASAP-seq 数据,因为这两种技术能够同时获取染色质可及性数据和基因表达数据 // We consider multimodal measurements profiling gene expression levels or chromatin accessibility simultaneously with surface protein levels, which can be obtained via CITE-seq and ASAP-seq.
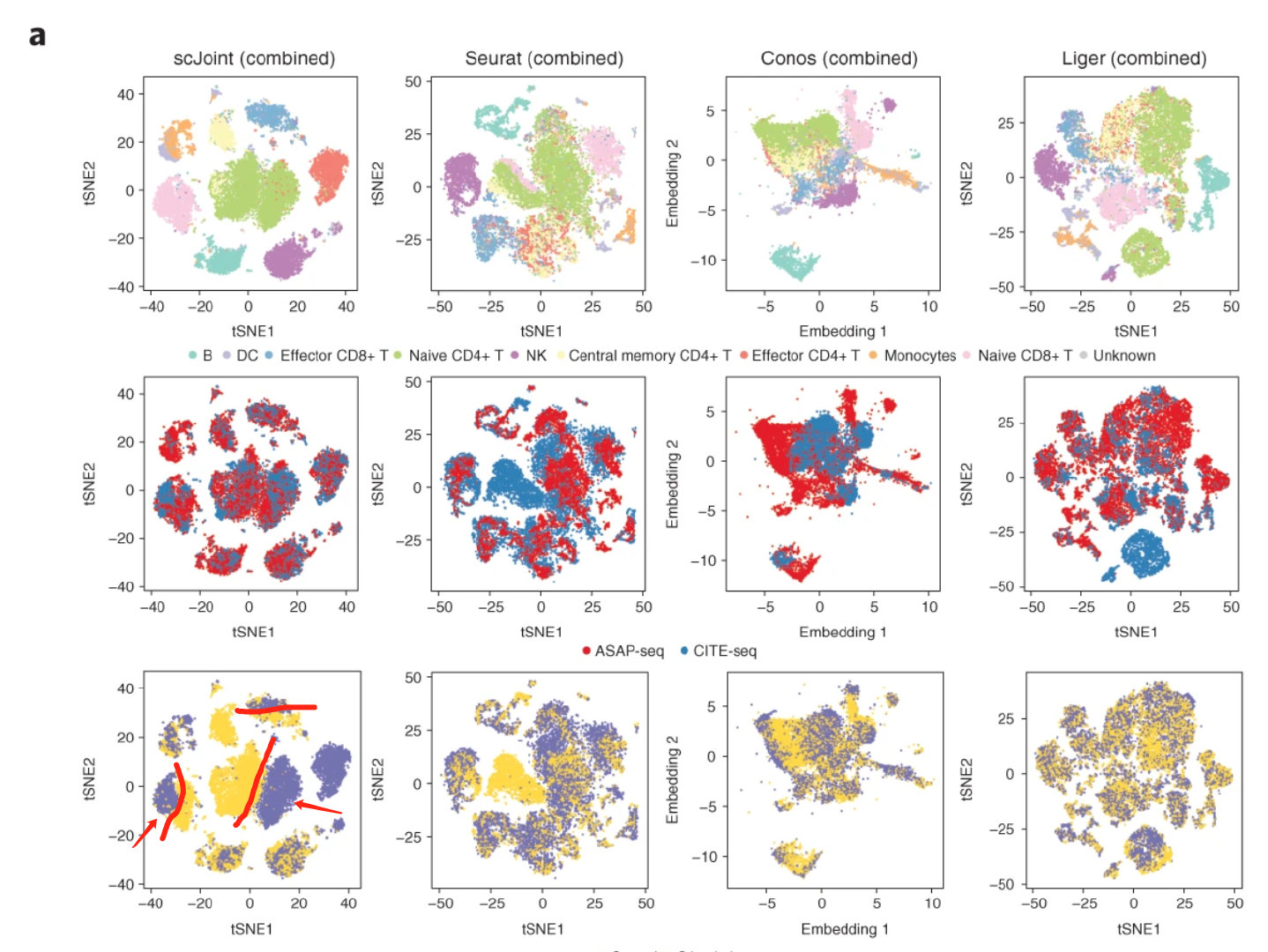
上面这幅图很好的展示了这个结果,很明显,各个细胞类型间能够明显的分群(第一行)。没有明显的测序方法差异(第二行)。在细胞群内,不同的生物学条件下具有较好的分类边界。
在文章的最后,作者将 scJoint 方法用于配对的数据,即在一个细胞中同时测scATAC-seq 和 scRNA-seq。然后根据基因表达水平对细胞进行标记,训练模型。随后将 scATAC-seq 数据用于对齐。准确率~70%。
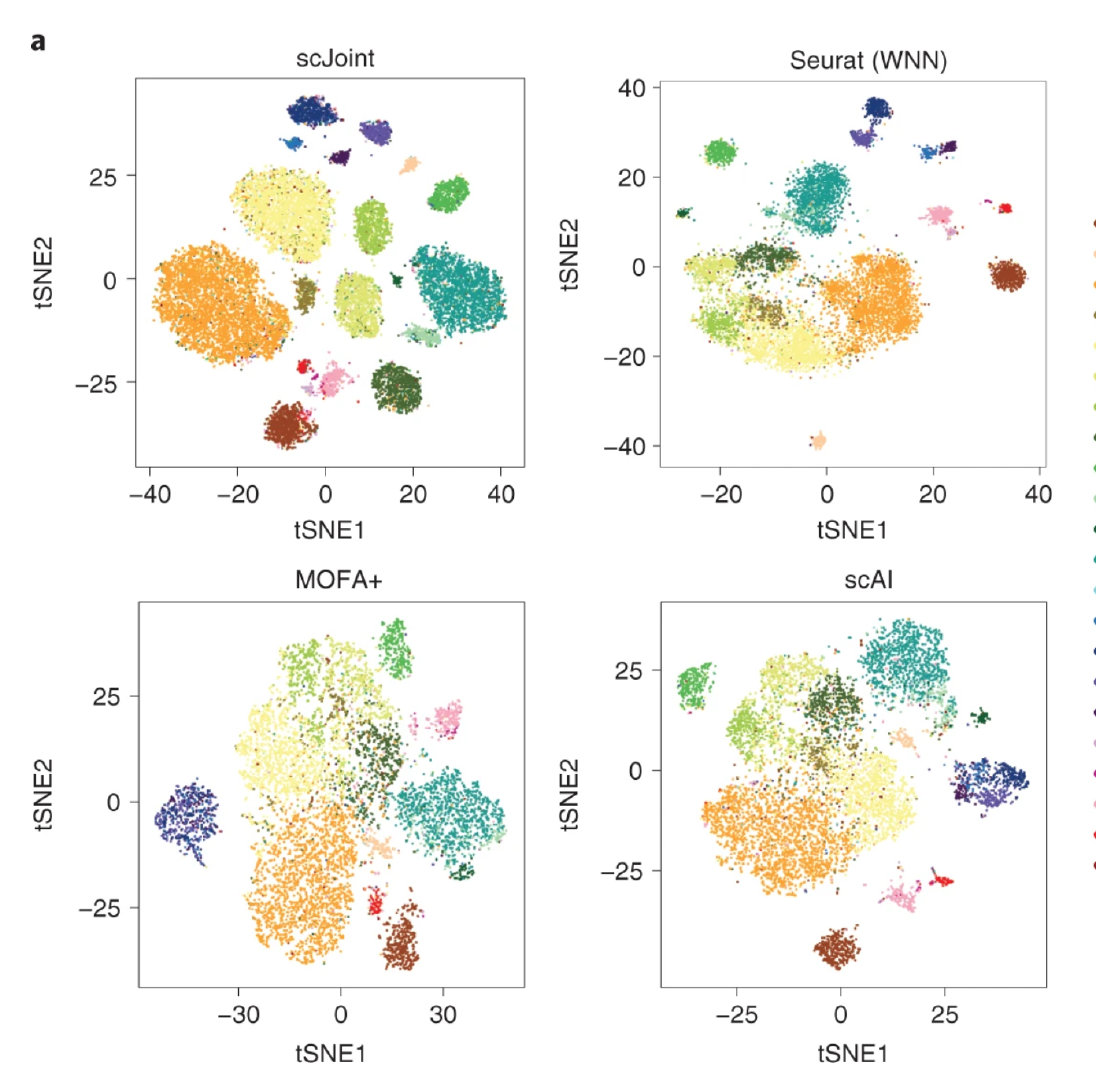
总结一下:
- 文章通过不同的角度证明 scJoint 在 ATAC 数据中的注释性能是具有很强的鲁棒性
- 相比于传统的方法 scJoint 具有更快的运行速度和更高的分辨率水平
最后,这里的话我个人有点小疑问:
如果说 RNA 表达水平与 ATAC 表达模式之间具有联系,且这种联系能够通过人工神经网络进行拟合的话。理论上配对的样本中应该有很好的结果,不应该只有 70%。是因为数据质量的问题吗?毕竟文章只评估了一套数据而已,所以很难下结论。
After advisement, if you still have questions, you can send me an E-mail asking for help
Best Regards,
Yuan.SH
---------------------------------------
please contact me via the following ways:
(a) E-mail: yuansh3354@gmail/163/outlook.com
(b) QQ: 1044532817
(c) WeChat: YuanSh181014
(d) Address: School of Basic Medical Sciences,
Fujian Medical University, Fuzhou,
Fujian 350108, China
---------------------------------------















 895
895











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









