







最近有小伙伴问道如下的文献图,简单明了的一种细胞一列进行marker gene表达展示。这个图其实就是展示了平均的细胞表达量而已。我们简单做一下。此外,顺便复现下基因注释。
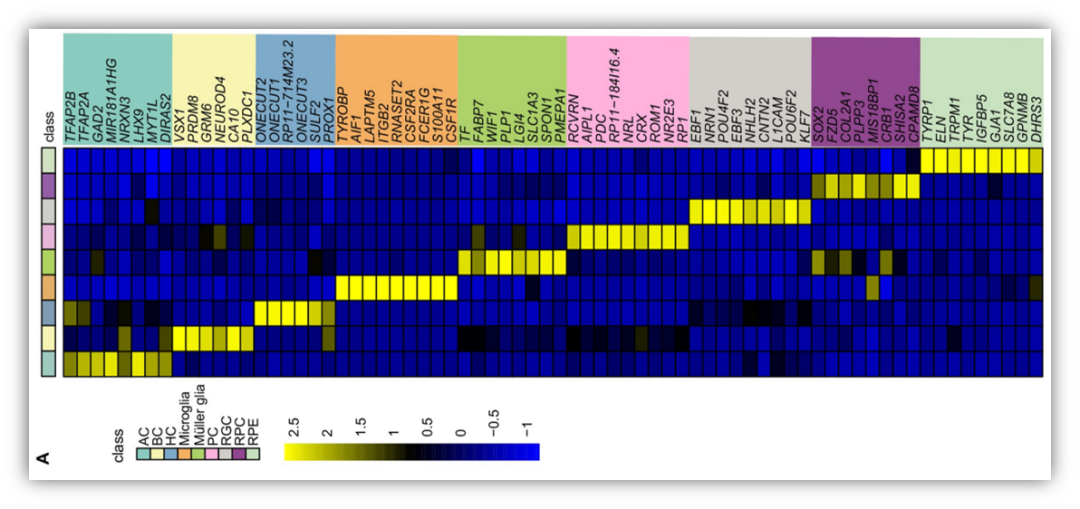
(reference:Dissecting the transcriptome landscape of the human fetal neural retina and retinal pigment epithelium by single-cell RNA-seq analysis)首先我们得到marker gene:
library(Seurat)
human_data <- readRDS("D:/KS/human_data.rds")
DefaultAssay(human_data) <- "RNA"
all.markers <- FindAllMarkers(human_data,
only.pos = TRUE,
min.pct = 0.25,
logfc.threshold = 0.75)
选择需要的marker gene进行展示,平均表达量使用seurat自带函数AverageExpression进行计算。热图使用Complexheatmap做即可。
#计算平均表达量
gene_cell_exp <- AverageExpression(human_data,
features = gene,
group.by = 'celltype',
slot = 'data')
gene_cell_exp <- as.data.frame(gene_cell_exp$RNA)
#complexheatmap作图
library(ComplexHeatmap)
#顶部细胞类型注释
df <- data.frame(colnames(gene_cell_exp))
colnames(df) <- 'class'
top_anno = HeatmapAnnotation(df = df,#细胞名/cluster
border = T,
show_annotation_name = F,
gp = gpar(col = 'black'),
col = list(class = c('Macrophage'="#9ECABE",
'T cell'="#F6F5B4",
'mDC'="#2F528F",
"Neutrophil"="#E3AD68",
"Mast"="#ACD45E")))#颜色设置
#数据标准化缩放一下
marker_exp <- t(scale(t(gene_cell_exp),scale = T,center = T))
Heatmap(marker_exp,
cluster_rows = F,
cluster_columns = F,
show_column_names = F,
show_row_names = T,
column_title = NULL,
heatmap_legend_param = list(
title=' '),
col = colorRampPalette(c("#0000EF","black","#FDFE00"))(100),
border = 'black',
rect_gp = gpar(col = "black", lwd = 1),
row_names_gp = gpar(fontsize = 10),
column_names_gp = gpar(fontsize = 10),
top_annotation = top_anno)

重点知识来了,为热图行名添加分屏注释:
col_cluster <- setNames(c(rep("#9ECABE",5), rep("#F6F5B4",6),
rep("#2F528F",5), rep("#E3AD68",5),
rep("#ACD45E",4)),
rownames(marker_exp))#设置对应标签颜色
row_info = rowAnnotation(foo = anno_text(rownames(marker_exp),
location = 0,
just = "left",
gp = gpar(fill = col_cluster,
col = "black"),
width = max_text_width(rownames(marker_exp))*1.2))
Heatmap(marker_exp,
cluster_rows = F,
cluster_columns = F,
show_column_names = F,
show_row_names = T,
column_title = NULL,
heatmap_legend_param = list(
title=' '),
col = colorRampPalette(c("#0000EF","black","#FDFE00"))(100),
border = 'black',
rect_gp = gpar(col = "black", lwd = 1),
row_names_gp = gpar(fontsize = 10),
column_names_gp = gpar(fontsize = 10),
top_annotation = top_anno)+row_info```
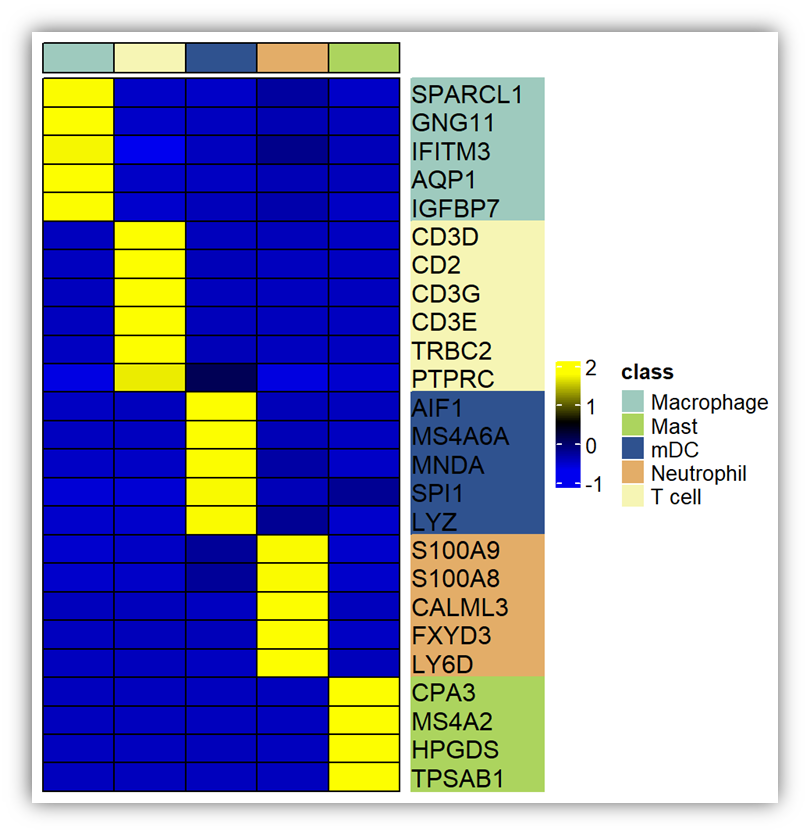
legend自己修饰一下就可以了。Complexheatmap真的很强大,有很多有用的功能,慢慢的我们探索。觉得分享有用的、点个赞、分享下再走呗!

















 1917
1917

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









