







今日目标:
(1)了解10X的数据的基本内容,我大概的看一下学姐发给我的这些数据都有啥,长什么样子的。毕竟fastq数据自己还是比较熟悉的。
(2)另外一方面,研究一下splitter的使用,其中有一些参数会根据你的数据的特征进行调整,我看一下主要的区别是什么?
(3)尝试修改splitter的代码,拆分数据(先有这个目标,虽然今天一天不一定可以搞定)。
(10:42)现在开始做这件事(之前都在摸鱼,不应该)。
1。10X Genomics的数据的基本格式
学姐给我的数据是这样的。

而我从10X Genomics数据库中下载的数据是这个样子的。

刚开始看的时候是有些懵的。
这里的R1/R2/I1的意思是什么?
- R1:cell barcode 和UMI序列
- R2:插入片段
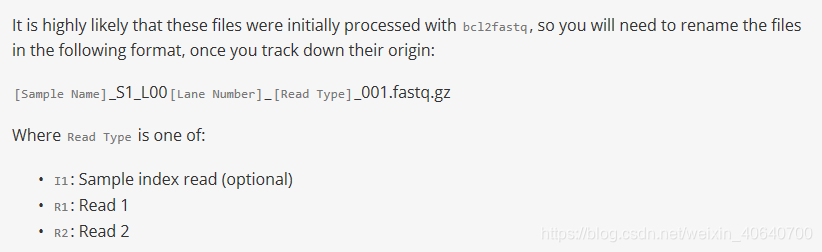

我们分别看一下read1和read2的内容。
(base) [xxzhang@mu02 pbmc4k]$ head pbmc4k_S1_L001_R1_001.fastq
@D000684:779:H53GNBCXY:1:1101:1198:2139 1:N:0:AAATGTGC
TNTCCTCGTTACCGATTGCATGCCCG
+
G#<GGGIIIIIIIIIIIIIIIIIIII
@D000684:779:H53GNBCXY:1:1101:1017:2155 1:N:0:AAATGTGC
AACTCCCCATTAGCCATGCGACGTGG
+
GGGGGIIIIIIIIIIIIGIIIIIGII
@D000684:779:H53GNBCXY:1:1101:1202:2159 1:N:0:AAATGTGC
AGAATAGAGACAAGCCGGTGACTATC
(base) [xxzhang@mu02 pbmc4k]$ head pbmc4k_S1_L001_R2_001.fastq
@D000684:779:H53GNBCXY:1:1101:1198:2139 2:N:0:AAATGTGC
AGATCTCTTGCACAGCATGATGAATATAGTTAATAAAACAGCATTGTACATTTCAAAATTGCCAAGTGTTAATTTCAAGT GTTCTCACCACAAAAATT
+
GGGGGIIIIIIIIIIIIIIIGIIIIIIIIIIGIIGIIGGIGGGIGIIGGIIIIGIIIGIIIIIIIIGGIIIIGIGIIIIG GGGGIIIIIGIIIIIGGA
@D000684:779:H53GNBCXY:1:1101:1017:2155 2:N:0:AAATGTGC
GNNNNNNNNNNNCNNNNNNNNNNNNNANNNNNNNCCNNNNNNGNNNNNNNTNCAGCCANANNNNNNNNNGNNNNNNNNNN NNNNNNNNNNNNNCNNAN
+
<###########<#############<#######<<######<#######<#<<GGGI#<#########<########## #############<##<#
@D000684:779:H53GNBCXY:1:1101:1202:2159 2:N:0:AAATGTGC
ATCTAATTAAACTAAAGAGCTTCTGCACAGCAAAAGAAACTATCATCAGAGTGAACAGGCAACCTACAGATTTGGAGAAA ATGTTTGCAATCTATCCA
暂时还看不懂是什么苗头。
2。10 Genomics的测序原理
上次简单的看了一下网上对于10X的测序原理的介绍。用费曼技巧总结的话,就是检验你输入的质量的方法,就是把你自己理解到的讲给别人听,看看别人是否能够听明白。那么,我现在就用我自己的话,对这个方法的主要流程进行总结。
(1)首先,形成一种油包水的结构。细胞和我们的barcode包在一起。存在唯一的对应关系。
(2)其次,在这个油包水的结构中,细胞破裂。mRNA得以释放出来。barcode序列旁边的ployT通过碱基互补配对的原则,与mRNA的3‘端的ployA区域结合。
(3)以mRNA为模板,从barcode-ployT开始进行cDNA的合成(5’–>3’)。cDNA合成之后,尾端加上已知的某种序列(用于下面双链的合成)。
(4)然后就是合成cDNA的双链。最后连接上P5,P7和index的序列(用于PCR的桥式扩增)。
(5)所以,我们最终用于测序的就是我们上面那张图展示的那一段。中间的inset区域,就是我们未知序列的mRNA的碱基信息。
那么通过这种方法,如何实现对一个细胞中表达什么基因(定性)以及表达多少这些信息(定量)的监测呢?
我觉得主要有以下几个方面。
(1)对于细胞的唯一性的标记主要是通过10X Barcode来实现的。首先Barcode的种类足够的多,能够实现唯一性的标记细胞。其次,在流式细胞仪中对于细胞与Barcode结合过程中流速的控制也是另外一方面。
(2)对于扩增的cDNA的唯一性主要是通过UMI的标记来实现的。之所以这样,是为了将PCR扩增产生的所有的子段都归在一起,避免PCR扩增的bias的现象。最终对基因的表达进行计数的时候,只需要统计UMI的种类即可。入下图所示。

3。了解scSplitter的参数需求
这里用于分割的工具主要还是QBRC的scSplitter这个工具。这个课题组还是挺有意思的,凡是应用范围比较广的,比较成熟的pipeline都写成了可以直接调用的工具,非常重视代码的复用。
https://github.com/zzhu33/scSplitter
看一下这个pipeline都有哪些的输入项。
- –f 输入文件名(txt格式,用tab键分割)
- –i 输入的fastq文件的路径
- –r 输出文件的路径
- –ind STAR索引存放的路径
- –chver 10X chemistry 版本
在这个list中有两个选择,一个是chemistry v1;一个是chemistry v2;但是关于这两者的主要区别,我不知道。 - –qc 默认值是lenient(宽松的)
这个是关于chemistry v1 配对的reads而言的QC mode;有“严格的”和“宽松的”两者选项。 - –cb 细胞barcode的长度 默认是16bp
- –sl sample index的长度 默认是8bp
- –ul UMI的长度 默认是10bp
- –tsol TSO的长度(这个要结合测序的过程而谈)对于老一点的chemistry v1配对的数据而言,是13bp
- –cc 细胞reads的cutoff值 对于一个细胞的barcode而言,最少要在多少条reads中找到,才说明这个barcode是有效的(也就是说这个细胞中有多少mRNA被标记成功,如果barcode很少,那其实不太有下面继续分析的价值了。另外虽然barcode唯一性的标记细胞,但是对于来源于同一个细胞中的mRNA的reads可以有很多条)默认值是3000
- –sc 样本reads的cutoff值 对于一个样本(一块组织,有很多的细胞)而言,最少有多少条reads来源于这个样本,我们认为是有效的。
- –ro 一组具有相同 UMI 的读数的中间对齐位置的最大偏差,以使读数有效(这个是在比对过程中的参数) 默认为500bp 在这个范围内 reads是有效的
- –po 对于配对的reads而言 中间对齐位置的最大偏差
- –so 选择是否保留所有合格的reads,还是每一种UMI只保留一个
- –ig 强制忽略样本index
- –u 在开始之前暂停程序用于让用户检查输入的参数是否正确
- –gz 是否压缩输出项
- –sam 是否保留sam文件用于比对的检查
简单的整理了一下。那我明白了。我现在需要弄清楚几个值(和师姐确认了一下)。
- 10X chemistry的版本:V1
- barcode 长度:16bp
- UMI长度:12bp
- TSO:13bp
- 这个数据是配对的数据
- 比对的参考基因组是hg38
- QC mode我们也选择宽松
(剩下的全部使用默认值,先跑一下看看,有点偷懒)
4。使用我们上面确定的参数,运行scSplitter
(1)首先准备txt文件。
R21010305-3-wenku-S1-3-wenku-S1_combined_R1.fastq.gz R21010305-3-wenku-S1-3-wenku-S1_combined_R2.fastq.gz
(2)准备指令。
python3 scSplitter.py --f inputNames.txt --i /home/xxzhang/workplace/QBRC/data/fastq/GEXst --r /home/xxzhang/workplace/QBRC/data/fastq/output --ind /home/xxzhang/workplace/QBRC/geneome/hg38 --chver 1 --qc strict --cb 16 --ul 12 --gz True --sam True
出错:
input directory: /home/xxzhang/workplace/QBRC/data/fastq/GEXst
the rows line is : [‘R21010305-3-wenku-S1-3-wenku-S1_combined_R1.fastq.gz’, ‘R21010305-3-wenku-S1-3-wenku-S1_combined_R2.fastq.gz’]
caution: one of the three read files (R1, R2, or I1) was not specified in inputNames
the run will proceed assuming I1 is not provided and all sample indices are presumed to be identical
output directory: /home/xxzhang/workplace/QBRC/data/fastq/output
Error -2: no genome index found at /home/xxzhang/workplace/QBRC/geneome/hg38!
才想起来,STAR比对的索引的文件夹应该是
/home/xxzhang/workplace/QBRC/geneome/hg38/STAR
所有重新的准备指令。
python3 scSplitter.py --f inputNames.txt --i /home/xxzhang/workplace/QBRC/data/fastq/GEXst --r /home/xxzhang/workplace/QBRC/data/fastq/output --ind /home/xxzhang/workplace/QBRC/geneome/hg38/STAR --chver 1 --qc strict --cb 16 --ul 12 --gz True --sam True --so True --u True
the rows line is : [‘R21010305-3-wenku-S1-3-wenku-S1_combined_R1.fastq.gz’, ‘R21010305-3-wenku-S1-3-wenku-S1_combined_R2.fastq.gz’]
caution: one of the three read files (R1, R2, or I1) was not specified in inputNames
the run will proceed assuming I1 is not provided and all sample indices are presumed to be identical
output directory: /home/xxzhang/workplace/QBRC/data/fastq/output
fastqs : R21010305-3-wenku-S1-3-wenku-S1_combined_R1.fastq.gz, R21010305-3-wenku-S1-3-wenku-S1_combined_R2.fastq.gz, …
STAR index : /home/xxzhang/workplace/QBRC/geneome/hg38/STAR
barcode_length : 16
sampl.ind.len : 8
UMI len : 12
cellReadsCutoff : 3000
sampleReadsCutoff : 1000000
maxReadOffset : 500
select one read per UMI : True
ignore sample indices : True
number of lanes : 1
user confirmation of inputs : True
compressed inputs : True
keep SAMs : True
version : 10x v1 chemistry paired
QC mode : strict
max pair offset : 50000000
82.156982421875 GB memory available, running with 20 processes
记录一下时间(19:46),看一下解压完成这1万个细胞需要多长时间(或者先解压部分,看看能不能用)。
屏幕显示的反馈的list:
finished sorting sams, 4476.066923618317 s, 11268.145693540573 s total
deleting intermediate files…
selecting reads based on alignment…
/home/xxzhang/miniconda3/lib/python3.6/multiprocessing/process.py:93: DtypeWarning: Columns (15) have mixed types.Specify dtype option on import or set low_memory=False.
self._target(*self._args, **self._kwargs)
multiprocessing.pool.RemoteTraceback:
“”"
Traceback (most recent call last):
File “/home/xxzhang/miniconda3/lib/python3.6/multiprocessing/pool.py”, line 119, in worker
result = (True, func(*args, **kwds))
File “/home/xxzhang/miniconda3/lib/python3.6/multiprocessing/pool.py”, line 47, in starmapstar
return list(itertools.starmap(args[0], args[1]))
File “/home/xxzhang/workplace/software/scSplitter/al_functions_m95.py”, line 448, in readSelection
selectl[i + (2 * medPairInd)] = True
UnboundLocalError: local variable ‘medPairInd’ referenced before assignment
“”"
The above exception was the direct cause of the following exception:
Traceback (most recent call last):
File “scSplitter.py”, line 496, in
main()
File “scSplitter.py”, line 447, in main
pool.starmap(af.readSelection, inputlist)
File “/home/xxzhang/miniconda3/lib/python3.6/multiprocessing/pool.py”, line 274, in starmap
return self._map_async(func, iterable, starmapstar, chunksize).get()
File “/home/xxzhang/miniconda3/lib/python3.6/multiprocessing/pool.py”, line 644, in get
raise self._value
UnboundLocalError: local variable ‘medPairInd’ referenced before assignment
以下,就是最后输出的结果(8/5 9:45)。每一个细胞对应两个fastq.gz的文件(因为是双端的数据)。所以,从目前看这个结果还是符合我们的预期的。

所以,昨天的目标也顺利的完成了。

















 3887
3887

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









