










前几天咱们分享了看完不会来揍我 | 孟德尔随机化万字长文详解(二)—— 代码实操 | 附代码注释 + 结果解读,很多小伙伴们反映在使用代码下载数据时会遇到各种网络或其他报错问题,令人头大的那种!不要慌!从数据库下载数据到本地的数据处理方法这就来啦!

-
如果小伙伴们有需求的话,可以加入我们的交流群:一定要知道 | 永久免费的环境友好型生信学习交流群又双叒叕来啦!| 伴随不定期群友好物分享!在这里,你可以稍有克制地畅所欲言!
-
超级建议大家在入群前或入群后可以看一下这个:干货满满 | 给生信小白的入门小建议 | 掏心掏肺版!绝对干货满满!让你不虚此看!
-
如果有需要个性化定制分析服务的小伙伴,可以看看这里:你要的个性化生信分析服务今天正式开启啦!定制你的专属解决方案!全程1v1答疑!!绝对包你满意!
直接开始!
数据要求
在进行**孟德尔随机化(Mendelian Randomization,MR)**分析时,关于曝露因子的 GWAS 数据,TwoSampleMR需要一个工具变量数据框,要求每行对应一个 SNP,至少需要 4 列最基本信息,包括:
- SNP - rsID,rsID 是 SNP 的唯一标识符。
- beta - 效应大小。如果是分类变量,我们就要使用log(OR),详见:看完不会来揍我 | 孟德尔随机化万字长文详解(二)—— 代码实操 | 附代码注释 + 结果解读。
- se - 效应大小的标准误差
- effect_allele - 效应等位基因
我们也可以提供以下对 MR 有用的其他信息:
- other_allele - 非效应等位基因
- eaf - 效应等位基因频率
- Phenotype - SNP具有效应的表型名称
我们还可以提供以下额外信息(非必须):
- chr - SNP 所在的染色体
- position - SNP 在染色体上的位置
- samplesize - 用于估计效应大小的样本大小
- ncase - 病例数量
- ncontrol - 对照组数量
- pval - SNP 与曝露因子关联的 P 值
- units - 以哪种单位呈现效应
- gene - SNP 的基因或其他注释
注意注意:不同来源的数据可能列名会有些许差异,大家要注意哈!
大家在下载完成后可以检查一下数据是否符合要求,从数据库下载的还好,一般不会有问题,主要是大家从相关文献获取数据的时候要注意这个问题。
从 IEU 数据库获取数据
数据下载
IEU 数据库官网:https://gwas.mrcieu.ac.uk/
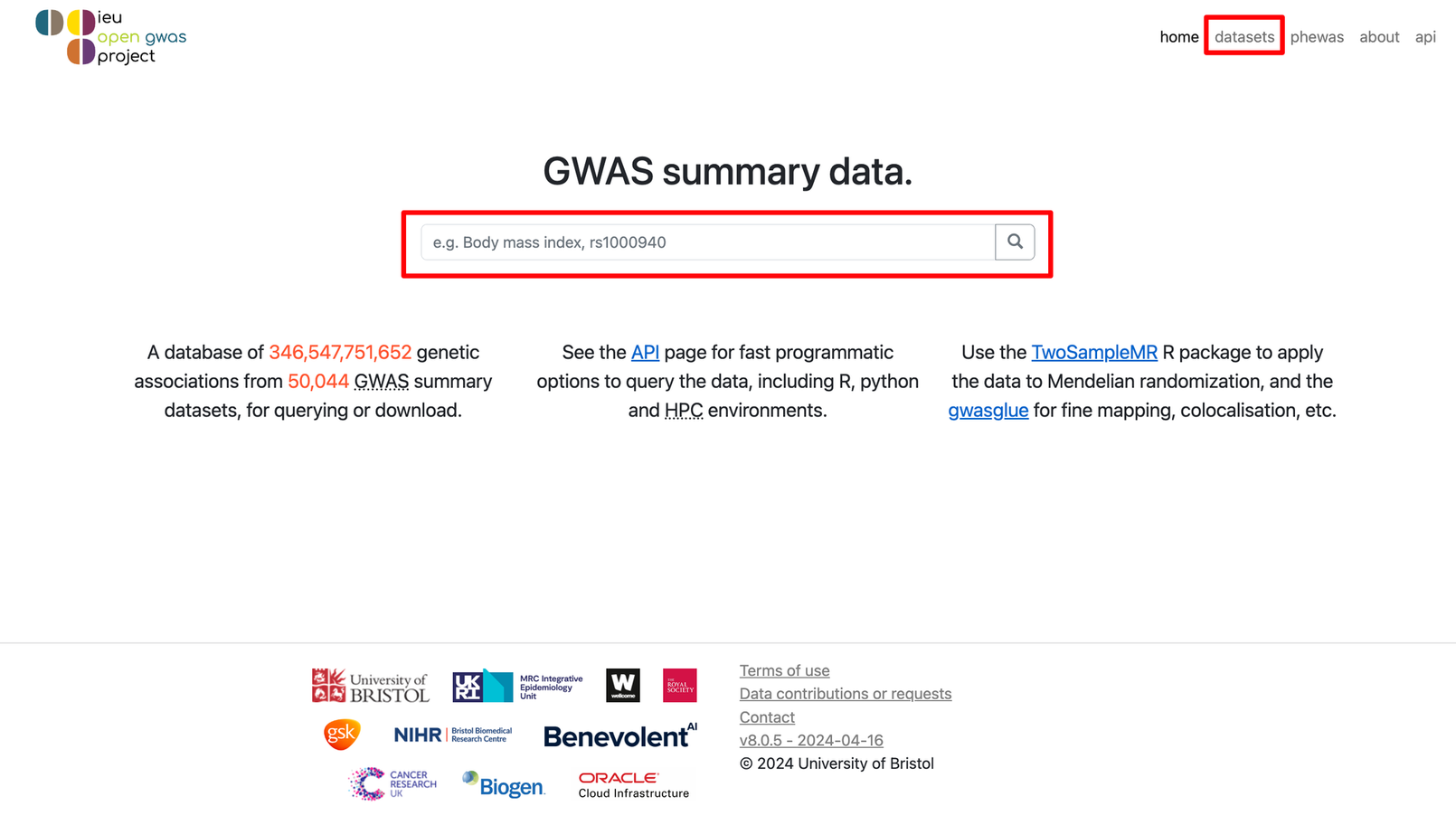
我们可以直接在中间的大框框里输入关键词,也可以点击右上角的datasets进入新的页面,在Trait contains的框框里输入关键词。比如我们这里就以body mass index(身体质量指数,也就是咱们常说的 BMI)作为关键词进行输入,然后点击🔍或者Filter,就会看到有很多数据被筛选出来啦!
我们今天就以探究 BMI(暴露)和乳腺癌(结局)之间的关系为例吧!
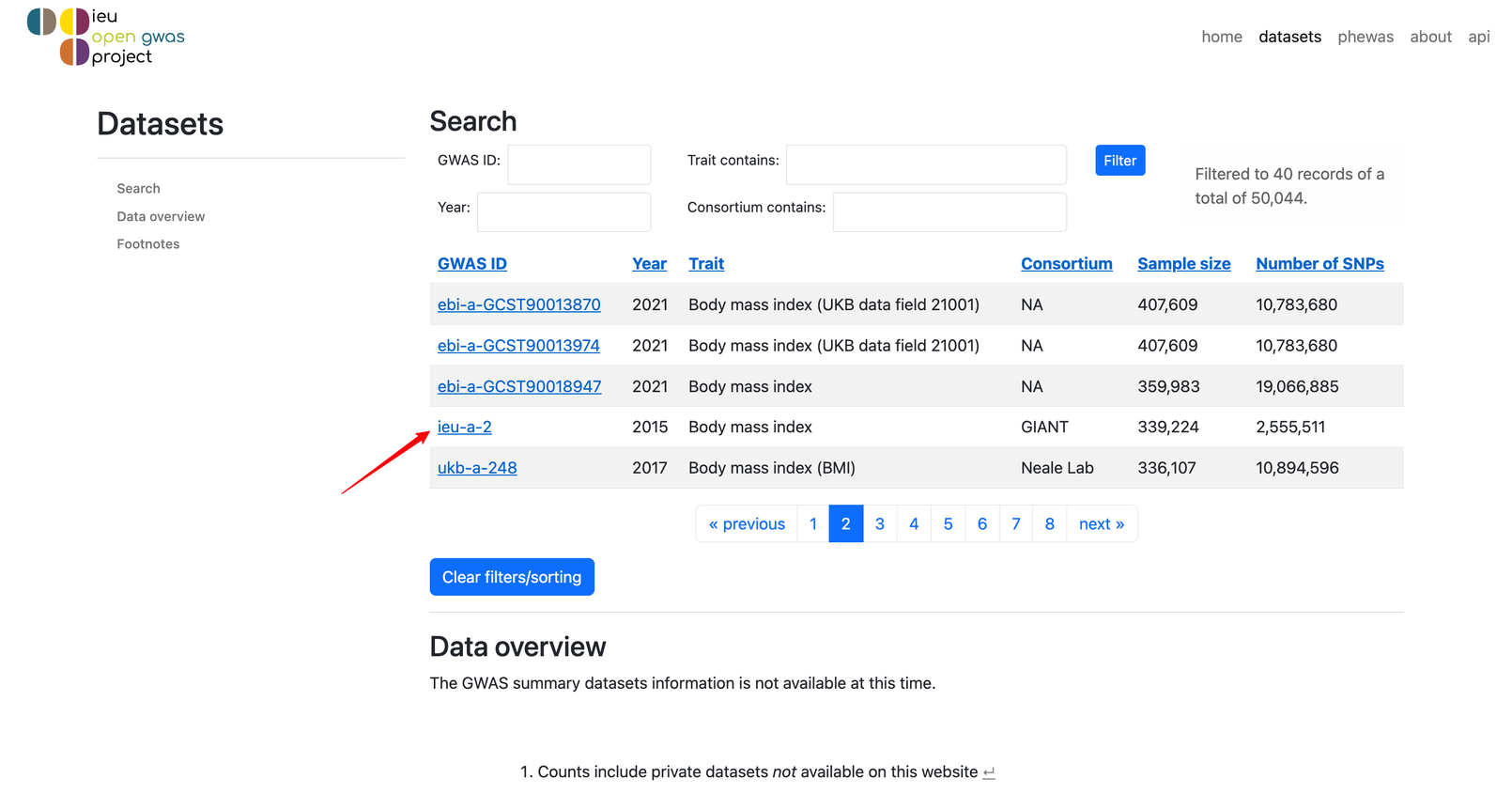
我们选择ieu-a-2这个数据吧!点进去!
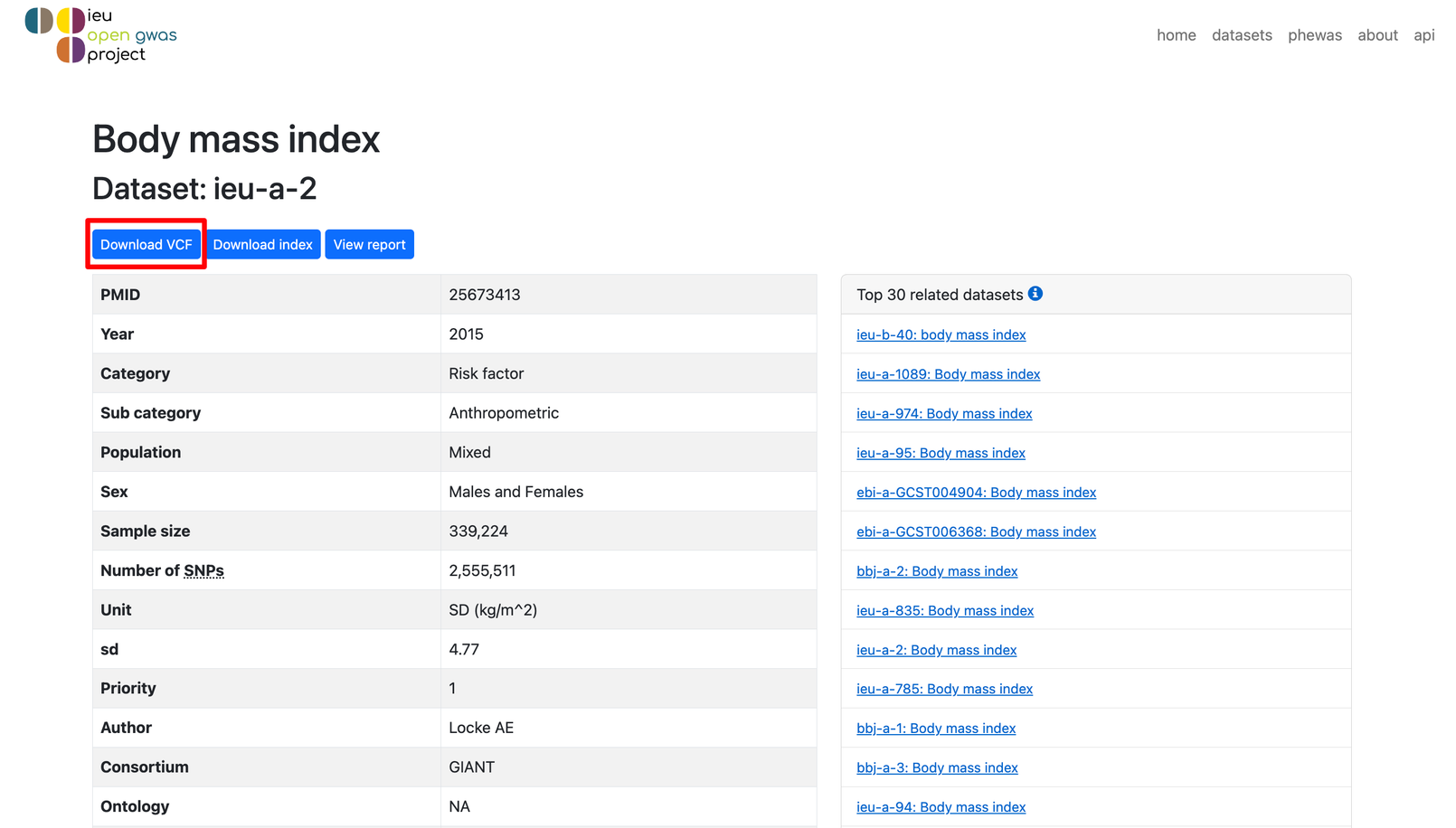
点击Download VCF(也可以用wget等命令直接下载,大家按自己习惯自由发挥!),即可下载完整的 GWAS 数据。
结局数据也是一样的下载方式,我这里就不演示了哈!我选择了ukb-b-16890(乳腺癌相关)这个数据。
下载完后长这样:

数据下载与处理
咱们开始读取它们,顺便处理!
# 加载包,没有的小伙伴记得安装一下哟!
# BiocManager::install("VariantAnnotation")
library(VariantAnnotation)
# remotes::install_github("mrcieu/gwasvcf")
library(gwasvcf)
# devtools::install_github("mrcieu/gwasglue")
library(gwasglue)
library(TwoSampleMR)
library(ieugwasr)
library(dplyr)
# 读取暴露数据,读取可能会有点慢,毕竟文件蛮大嘛,大家不要着急哈,这是正常滴!
exposure_vcf <- readVcf("./ieu-a-2.vcf.gz")
# 我们看看这个文件里面都是什么
exposure_vcf
# class: CollapsedVCF
# dim: 2554285 1
# rowRanges(vcf):
# GRanges with 5 metadata columns: paramRangeID, REF, ALT, QUAL, FILTER
# info(vcf):
# DataFrame with 1 column: AF
# info(header(vcf)):
# Number Type Description
# AF A Float Allele Frequency
# geno(vcf):
# List of length 9: ES, SE, LP, AF, SS, EZ, SI, NC, ID
# geno(header(vcf)):
# Number Type Description
# ES A Float Effect size estimate relative to the alternative allele
# SE A Float Standard error of effect size estimate
# LP A Float -log10 p-value for effect estimate
# AF A Float Alternate allele frequency in the association study
# SS A Float Sample size used to estimate genetic effect
# EZ A Float Z-score provided if it was used to derive the EFFECT and SE fields
# SI A Float Accuracy score of summary data imputation
# NC A Float Number of cases used to estimate genetic effect
# ID 1 String Study variant identifier
小小解读一下下:
- class:
CollapsedVCF表示这是一个 VCF 文件的压缩版本。 - dim:
2554285 1表示该 VCF 文件包含 2336260 个行(即 SNP)和 1 个样本。- rowRanges(vcf): 该部分包含 5 个元数据列,分别是
paramRangeID,REF,ALT,QUAL,FILTER。这些列提供了关于每个 SNP 的基本信息。
- rowRanges(vcf): 该部分包含 5 个元数据列,分别是
- info(vcf): 包含一个 DataFrame,列名为
AF,表示Allele Frequency(等位基因频率)。 - info(header(vcf)): 提供了
AF列的描述,它是一个浮点数,代表等位基因的频率。 - geno(vcf): 这是一个长度为 9 的列表,包含各种遗传效应和统计量的估计值。
- ES: 效应大小相对于替代等位基因的估计值。
- SE: 效应大小估计的标准误差。
- LP: 效应估计的-log10 P值。
- AF: 关联研究中的替代等位基因频率。
- SS: 用于估计遗传效应的样本大小。
- EZ: 如果用于推导
EFFECT和SE字段,则提供的Z分数。 - SI: 摘要数据插补的准确度分数。
- NC: 用于估计遗传效应的病例数量。
- ID: SNP 标识符。
接下来我们依据 P 值进行过滤,并将其转换为TwoSampleMR需要的格式,顺便去除一下连锁不平衡。
# 过滤 P 值 —— 说实话,过滤 P 值这个步骤呐,放在哪一步都可以,大家随意!
exposure_vcf_p_filter <- query_gwas(vcf = exposure_vcf, pval = 5e-08)
# 转换为 TwoSampleMR 需要的格式
exposure_data <- gwasvcf_to_TwoSampleMR(exposure_vcf_p_filter)
head(exposure_data)[1:4, ]
# chr.exposure pos.exposure other_allele.exposure effect_allele.exposure beta.exposure
# 1 1 47684677 T G -0.0168
# 2 1 47690438 G T 0.0165
# 3 1 49376820 T C 0.0209
# 4 1 49438005 G A -0.0225
# se.exposure pval.exposure eaf.exposure samplesize.exposure ncase.exposure SNP
# 1 0.0030 2.181976e-08 0.5333 339152 NA rs977747
# 2 0.0030 3.416017e-08 0.4746 338820 NA rs2984618
# 3 0.0035 2.371974e-09 0.2250 339110 NA rs1343424
# 4 0.0031 3.773115e-13 0.6333 338453 NA rs3127553
# ncontrol.exposure exposure mr_keep.exposure pval_origin.exposure id.exposure
# 1 NA ieu-a-2 TRUE reported FkMOXE
# 2 NA ieu-a-2 TRUE reported FkMOXE
# 3 NA ieu-a-2 TRUE reported FkMOXE
# 4 NA ieu-a-2 TRUE reported FkMOXE
# 可以看到和咱们之前用 extract_instruments() 下载的数据格式是一致的!棒棒哒!
# 去除连锁不平衡(有些小伙伴是不是网不好!不止服务器崩溃!自己也崩溃!不要慌!)
exposure_data_clumped <- clump_data(exposure_data, # 暴露数据
clump_kb = 10000, # clumping 的窗口大小
clump_r2 = 0.001, # clumping 的 r^2 阈值
pop = "EUR") # 用于参考的人种(默认为“EUR/欧洲人”,具体大家可以去问号一下函数)
# Please look at vignettes for options on running this locally if you need to run many instances of this command.
# Clumping 6UhrjN, 2041 variants, using EUR population reference
# Error in api_query("ld/clump", query = list(rsid = dat[["rsid"]], pval = dat[["pval"]], :
# The query to MR-Base exceeded 300 seconds and timed out. Please simplify the query
哎!报错啦!超时啦!
是不是很多小伙伴好不容易搞定了第一步,在这里又卡在了这一步!不要慌!咱们就纯纯本地来!
不知道为啥,也是蛮巧的,我之前跑就没有这个问题,不知道为啥今天一直是这样,一定是服务器为了方便我给大家展示特地把自己搞成这样的哈哈哈哈哈哈哈!
说真的,我在一开始运行这步的时候还没有这个报错,但是有更玄幻更离谱的事情发生,就是就是,它居然给我筛选的只剩0个SNP了,特诡异!本来以为是我的数据问题,然后随便找个别人的教程,用了和它一样的数据,具体我还是0!完全无法理解,后来我又去各种检查源代码各种修改各种折腾,居然还是不行!玄学!离谱!给我气了一天!哎!不知道你们如果成功运行的话,会不会出现这种情况,哎!没有更好!咱就方便多了,如果遇到和我一样离谱的情况的话,就只能按照我下面介绍的方法啦!也不算特别麻烦,就是需要辛苦大家下载一下那个巨大的数据罢了!加油!祝大家各种顺利!
首先,咱们要下载连锁不平衡的参考数据集到本地,下载地址为:http://fileserve.mrcieu.ac.uk/ld/1kg.v3.tgz(有点大,大家慢慢来,哎)
下载后解压,解压命令参考(大家可以自行选择自己最最最喜欢的解压方式):
tar zxvf 1kg.v3.tgz
里面包含这些数据:
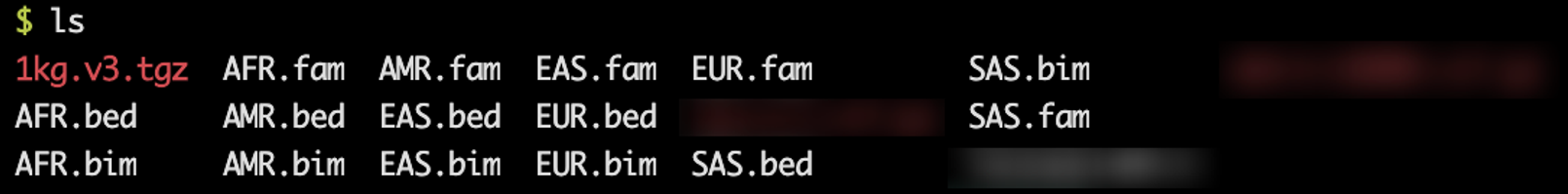
好多人种,咱们今天的数据集是欧洲人,所以咱们选择参考人种为欧洲人,那就会用到EUR.bed、EUR.bim、EUR.fam这三个文件。
其次,咱们还要下载一个叫plink的二进制文件,下载地址为:https://github.com/MRCIEU/genetics.binaRies/tree/master/binaries
可以看到有Linux版本,也有Windows版本,大家点进去直接下载适合自己的就好啦!比如我需要Linux版本的,那我就这样这样那样那样哈哈哈,按照下图所示挨个点击箭头区域即可(简单粗暴):

好,下载完plink 执行文件后,咱们就可以开始进行本地去除连锁不平衡操作啦!
# 使用 ld_clump() 函数进行 clumping,也就是去除连锁不平衡(本地本地本地)
exposure_data_clumped <- ld_clump(
# 指定包含`rsid`、`pval`和`id`的数据框
dat = tibble(rsid = exposure_data$SNP, # 从 exposure_data 中提取 SNP 标识符
pval = exposure_data$pval.exposure, # 从 exposure_data 中提取关联 p 值
id = exposure_data$exposure), # 从 exposure_data 中提取 SNP 的 id
clump_kb = 10000, # 设置 clumping 窗口大小
clump_r2 = 0.001, # 指定用于 LD 的 r^2 阈值
clump_p = 1, # 设置 p 值阈值为 1,这样所有的 SNP 都会被纳入 clump,因为咱们前面已经进行过 p 值过滤了嘛
bfile = "./EUR", # 指定 LD 的参考数据集,注意修改你的路径
plink_bin = "./plink" # 使用指定目录下的 plink 二进制文件,注意修改你的路径
)
# 大家在进行这一步的时候,可能会遇到如下报错:
# Clumping ieu-a-2, 2041 variants, using EUR population reference
# sh: 1: ./plink: Permission denied
# Error in file(file, "rt") : cannot open the connection
# In addition: Warning message:
# In file(file, "rt") :
# cannot open file '/tmp/RtmpRXcyTx/file25501f4f6efa35.clumped': No such file or directory
哎嘿,又报错了!那这又是为啥捏!
这意味着你没有足够的权限运行 plink。咱们需要给 plink 文件添加执行权限,在终端,也就是输入 Linux 命令的那个窗口,导航到plink所在目录,咱们运行下面这行命令(注意修改路径哈):
chmod +x /这里是/你的/plink文件/所在路径/MendelianRandomization/plink
这样,咱们就给plink添加了执行权限。
然后还是运行上面那段代码:
# 使用 ld_clump() 函数进行 clumping,也就是去除连锁不平衡(本地本地本地)
exposure_data_clumped <- ld_clump(
# 指定包含`rsid`、`pval`和`id`的数据框
dat = tibble(rsid = exposure_data$SNP, # 从 exposure_data 中提取 SNP 标识符
pval = exposure_data$pval.exposure, # 从 exposure_data 中提取关联 p 值
id = exposure_data$exposure), # 从 exposure_data 中提取 SNP 的 id
clump_kb = 10000, # 设置 clumping 窗口大小
clump_r2 = 0.001, # 指定用于 LD 的 r^2 阈值
clump_p = 1, # 设置 p 值阈值为 1,这样所有的 SNP 都会被纳入 clump,因为咱们前面已经进行过 p 值过滤了嘛
bfile = "./EUR", # 指定 LD 的参考数据集,注意修改你的路径
plink_bin = "./plink" # 使用指定目录下的 plink 二进制文件,注意修改你的路径
)
# Clumping ieu-a-2, 2041 variants, using EUR population reference
# PLINK v1.90b6.21 64-bit (19 Oct 2020) www.cog-genomics.org/plink/1.9/
# (C) 2005-2020 Shaun Purcell, Christopher Chang GNU General Public License v3
# Logging to /tmp/RtmpRXcyTx/file25501f72c90730.log.
# Options in effect:
# --bfile ./EUR
# --clump /tmp/RtmpRXcyTx/file25501f72c90730
# --clump-kb 10000
# --clump-p1 1
# --clump-r2 0.001
# --out /tmp/RtmpRXcyTx/file25501f72c90730
#
# 515586 MB RAM detected; reserving 257793 MB for main workspace.
# 8550156 variants loaded from .bim file.
# 503 people (0 males, 0 females, 503 ambiguous) loaded from .fam.
# Ambiguous sex IDs written to /tmp/RtmpRXcyTx/file25501f72c90730.nosex .
# Using 1 thread (no multithreaded calculations invoked).
# Before main variant filters, 503 founders and 0 nonfounders present.
# Calculating allele frequencies... done.
# 8550156 variants and 503 people pass filters and QC.
# Note: No phenotypes present.
# Warning: 'rs9930333' is missing from the main dataset, and is a top variant.
# Warning: 'rs8083289' is missing from the main dataset, and is a top variant.
# Warning: 'rs2683992' is missing from the main dataset, and is a top variant.
# 27 more top variant IDs missing; see log file.
# --clump: 78 clumps formed from 2011 top variants.
# Results written to /tmp/RtmpRXcyTx/file25501f72c90730.clumped .
# Removing 1963 of 2041 variants due to LD with other variants or absence from LD reference panel
这次运行成功了吧!!!
输出包括 PLINK 的版本信息和版权信息、logging 信息、运行选项、系统信息、数据加载信息(共有 8550156 个变异和 503 个人被加载)、警告信息(一些SNPs在主数据集中缺失)和 clumping 结果等,我们最需要关注的就是:它还剩 78 个 SNP!
# 查看去除连锁不平衡后的数据
head(exposure_data_clumped)
# # A tibble: 6 × 3
# rsid pval id
# <chr> <dbl> <chr>
# 1 rs977747 2.18e- 8 ieu-a-2
# 2 rs657452 2.12e-13 ieu-a-2
# 3 rs7531118 1.88e-28 ieu-a-2
# 4 rs17381664 4.57e-11 ieu-a-2
# 5 rs11165643 1.43e-13 ieu-a-2
# 6 rs7550711 5.06e-14 ieu-a-2
# 可以看到它只有 3 列,不慌!咱把它和前面的数据整合一下就好!
exposure_data_clumped <- exposure_data %>% filter(exposure_data$SNP %in% exposure_data_clumped$rsid)
# 再看看数据长啥样
head(exposure_data_clumped)
# chr.exposure pos.exposure other_allele.exposure effect_allele.exposure beta.exposure se.exposure pval.exposure eaf.exposure
# 1 1 47684677 T G -0.0168 0.0030 2.181976e-08 0.5333
# 2 1 49589847 A G -0.0227 0.0031 2.123244e-13 0.5833
# 3 1 72837239 T C 0.0331 0.0030 1.880182e-28 0.6083
# 4 1 78048331 T C 0.0201 0.0031 4.567726e-11 0.4250
# 5 1 96924097 C T 0.0221 0.0030 1.433838e-13 0.5750
# 6 1 110082886 C T 0.0659 0.0087 5.059411e-14 0.0339
# samplesize.exposure ncase.exposure SNP ncontrol.exposure exposure mr_keep.exposure pval_origin.exposure id.exposure
# 1 339152 NA rs977747 NA ieu-a-2 TRUE reported 6UhrjN
# 2 318585 NA rs657452 NA ieu-a-2 TRUE reported 6UhrjN
# 3 338123 NA rs7531118 NA ieu-a-2 TRUE reported 6UhrjN
# 4 339065 NA rs17381664 NA ieu-a-2 TRUE reported 6UhrjN
# 5 337797 NA rs11165643 NA ieu-a-2 TRUE reported 6UhrjN
# 6 313621 NA rs7550711 NA ieu-a-2 TRUE reported 6UhrjN
# 哎嘿,齐活啦!
那到现在为止,咱们暴露数据就算是搞定啦!接下来是结局数据,步骤和暴露数据完全一样,我们就快快搞一下!
在之前在看完不会来揍我 | 孟德尔随机化万字长文详解(二)—— 代码实操 | 附代码注释 + 结果解读中,我们是用extract_outcome_data函数从暴露数据中挑选结局相关数据,其实就是取交集获取共有的 SNP,那我们这里,也对暴露数据和结局数据取交集!
# 读取结局数据,读取可能会有点慢,毕竟文件蛮大嘛,大家不要着急哈,这是正常滴!
outcome_vcf <- readVcf("./ukb-b-16890.vcf.gz")
# 转换为 TwoSampleMR 需要的格式
outcome_data <- gwasvcf_to_TwoSampleMR(outcome_vcf)
# 结局数据与暴露数据取交集,获取共有 SNP
data_common <- merge(exposure_data_clumped, outcome_data, by = "SNP")
dim(data_common)
# [1] 77 31
# 可以看到最终还剩 77 个 SNP
# 看一下有哪些列,放在这里便于后面的格式转换,主要是方便看
colnames(data_common)
# [1] "chr.exposure" "pos.exposure" "other_allele.exposure"
# [4] "effect_allele.exposure" "beta.exposure" "se.exposure"
# [7] "pval.exposure" "eaf.exposure" "samplesize.exposure"
# [10] "ncase.exposure" "SNP" "ncontrol.exposure"
# [13] "exposure" "mr_keep.exposure" "pval_origin.exposure"
# [16] "id.exposure"
# 提取结局数据,并进行格式修改,以用于后续的 MR 分析
outcome_data <- format_data(outcome_data,
# 设置类型为 "outcome",表示这是一个结局数据
type = "outcome",
# 选择用于 MR 分析的 SNP,这里使用了 data_common 数据集中的 SNP 列
snps = data_common$SNP,
# 设置 SNP 列的名称
snp_col = "SNP",
# 设置效应大小 (beta) 的列名称
beta_col = "beta.exposure",
# 设置标准误差 (SE) 的列名称
se_col = "se.exposure",
# 设置效应等位基因频率 (EAF) 的列名称
eaf_col = "eaf.exposure",
# 设置效应等位基因的列名称
effect_allele_col = "effect_allele.exposure",
# 设置非效应等位基因的列名称
other_allele_col = "other_allele.exposure",
# 设置 p 值 (p-value) 的列名称
pval_col = "pval.exposure",
# 设置样本大小 (sample size) 的列名称
samplesize_col = "samplesize.exposure",
# 设置病例数 (number of cases) 的列名称
ncase_col = "ncase.exposure",
# 设置 id 的列名称
id_col = "id.exposure")
head(outcome_data)
# other_allele.outcome effect_allele.outcome beta.outcome se.outcome pval.outcome eaf.outcome
# 1 T G 2.06895e-04 0.000308588 0.50000000 0.584698
# 2 A G 5.98540e-04 0.000312550 0.05499966 0.608716
# 3 T C -3.21544e-04 0.000306840 0.29000000 0.531137
# 4 T C 8.35377e-05 0.000308980 0.78999983 0.407134
# 5 C T 3.08316e-04 0.000308869 0.32000002 0.590060
# 6 A G -2.52780e-04 0.000376396 0.50000000 0.205203
# samplesize.outcome ncase.outcome SNP id.outcome outcome mr_keep.outcome pval_origin.outcome
# 1 462933 10303 rs977747 IT2c0O outcome TRUE reported
# 2 462933 10303 rs657452 IT2c0O outcome TRUE reported
# 3 462933 10303 rs7531118 IT2c0O outcome TRUE reported
# 4 462933 10303 rs17381664 IT2c0O outcome TRUE reported
# 5 462933 10303 rs11165643 IT2c0O outcome TRUE reported
# 6 462933 10303 rs543874 IT2c0O outcome TRUE reported
# 可以看到也和之前代码直接下载的数据是一样滴!
MR 分析正式开始
后面的分析步骤和之前介绍的完全一样!咱们这里简单展示一下基本流程,关于具体细节介绍、参数调整、结果解读等等,详见:看完不会来揍我 | 孟德尔随机化万字长文详解(二)—— 代码实操 | 附代码注释 + 结果解读
# MR 分析正式开始
# 整合暴露数据和结局数据
data <- harmonise_data(
exposure_dat = exposure_data_clumped, # 指定暴露数据,这是之前从 GWAS 数据集中提取的工具变量数据
outcome_dat = outcome_data # 指定结局数据,这是从另一个 GWAS 数据集中提取的关联结局数据
)
# Harmonising ieu-a-2 (6UhrjN) and outcome (IT2c0O)
# 噢对,我们也可以修改一下暴露因素和结局因素的名称,方便人类肉眼阅读大脑理解
exposure_data_clumped$id.exposure <- "BMI"
outcome_data$id.outcome <- "BRCA"
# 再整合
data <- harmonise_data(
exposure_dat = exposure_data_clumped, # 指定暴露数据,这是之前从 GWAS 数据集中提取的工具变量数据
outcome_dat = outcome_data # 指定结局数据,这是从另一个 GWAS 数据集中提取的关联结局数据
)
# Harmonising ieu-a-2 (BMI) and outcome (BRCA)
# MR 分析
result <- mr(data)
# 异质性检验
mr_heterogeneity(data)
# 水平多效性检验
mr_pleiotropy_test(data)
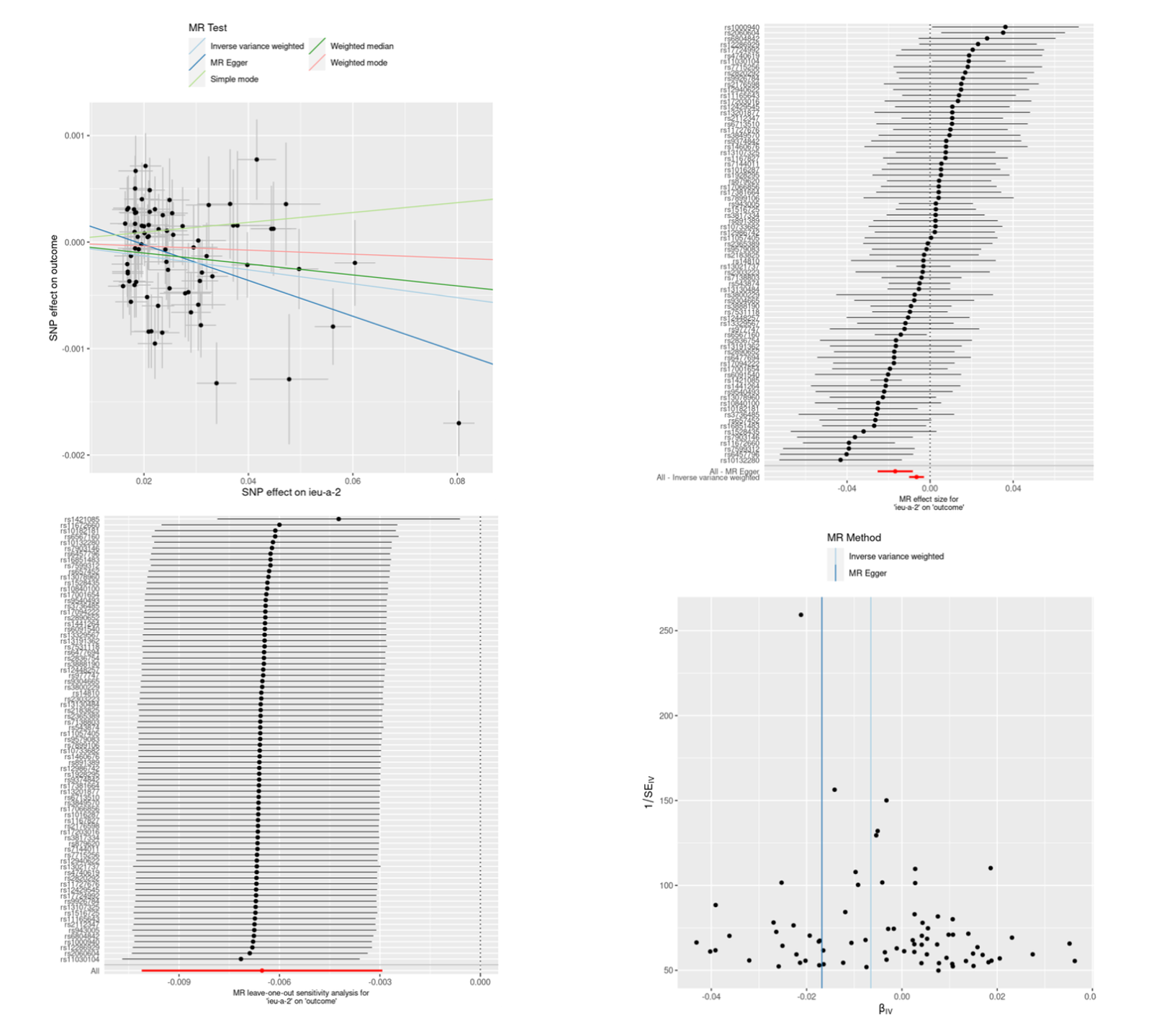
# 散点图
p1 <- mr_scatter_plot(result, data)
p1
# 森林图
result_single <- mr_singlesnp(data)
p2 <- mr_forest_plot(result_single)
p2
# 留一图
result_loo <- mr_leaveoneout(data)
p3 <- mr_leaveoneout_plot(result_loo)
p3
# 漏斗图
result_single <- mr_singlesnp(data)
p4 <- mr_funnel_plot(result_single)
p4
文末碎碎念
那今天的分享就到这里啦!我们下期再见哟!
最后顺便给自己推荐一下嘿嘿嘿!
如果我的分享对你有用的话,欢迎关注点赞在看转发分享阿巴阿巴阿巴阿巴巴巴!这可是我的第一原动力!
蟹蟹你们的喜欢和支持!!!
参考资料
- https://mrcieu.github.io/TwoSampleMR/articles/exposure.html
- https://mrcieu.github.io/TwoSampleMR/articles/outcome.html
- https://github.com/ixxmu/mp_duty/issues/4670
- https://www.bilibili.com/video/BV1LV4y1b7v5
- https://github.com/MRCIEU/gwasvcf
- https://github.com/MRCIEU/gwasglue


















 2551
2551

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











