










01 背景
TransDecoder(在转录本中查找编码区域),寻找ORF区域。ORF是“开放阅读框架”的缩写,它指的是在核酸序列(如DNA或RNA)中,从起始密码子(通常是ATG,编码甲硫氨酸)到终止密码子(例如TAA,TAG或TGA)之间的连续序列,它们可以被翻译成蛋白质。ORF的长度通常以碱基对或氨基酸数量来度量,它们在基因组学和蛋白质组学中被用来预测可能的编码序列。
1.1 ORF与CDS
ORF(开放阅读框架)和CDS(编码序列)之间有一些区别:
1. ORF(开放阅读框架):
- ORF是核酸序列中的一段连续序列,从起始密码子到终止密码子,可以被翻译成蛋白质。
- ORF的长度通常以碱基对或氨基酸数量来衡量。
- 它是在基因组或转录本水平上识别的可能的编码序列。
2. CDS(编码序列):
- CDS是完整的编码蛋白质的序列,即包含在mRNA中,从起始密码子到终止密码子之间的序列。
- CDS通常是在已知基因或转录本中定义的,而不是在全基因组水平上进行预测。
- 它是已知或已注释的编码蛋白质的实际序列。
总的来说,ORF是在基因组或转录本水平上预测的可能的编码序列,而CDS是已知或已注释的真实编码蛋白质的序列。
1.2 TransDecoder功能实现
TransDecoder可以识别转录本序列中的候选编码区域,例如由Trinity生成的de novo RNA-Seq转录组装序列,或者基于RNA-Seq对基因组的序列比对构建的序列。
TransDecoder适用于整个单一生物体的转录组,涉及数千个转录本序列作为输入。
如果您提供少量序列作为输入,TransDecoder可能无法正常工作,因为它需要基于从输入中派生的数百个候选项来训练一个物种特异性模型。
TransDecoder根据以下标准确定可能的编码序列:
- 在转录本序列中找到最小长度的开放阅读框架(ORF)。
- 计算与GeneID软件类似的对数似然分数 > 0。
- 当在第一个读框架中评分时,上述编码分数最高,与其他两个正向读框架中的分数相比。
- 如果发现一个候选ORF完全被另一个候选ORF的坐标所包含,则报告较长的那个。然而,单个转录本可以报告多个ORF(允许操纵子,嵌合体等)。
- 构建/训练/使用PSSM来优化起始密码子预测。
- 可选的,推测的肽段与Pfam域的匹配超过噪声截止分数。
02 参考
https://github.com/TransDecoder/TransDecoder #官网03 安装
wget -c https://github.com/TransDecoder/TransDecoder/archive/refs/tags/TransDecoder-v5.7.1.tar.gz #下载
tar -zxvf TransDecoder-v5.7.1.tar.gz #解压
mv TransDecoder-TransDecoder-v5.7.1 TransDecoder-v5.7.1 #修改名字04 使用
#1 第一步
./TransDecoder.LongOrfs -h
Translation:
Transdecoder.LongOrfs - 转录组蛋白质预测
必需项:
-t <string> 文件名为 transcripts.fasta 的转录本
可选项:
--gene_trans_map <string> 基因到转录本标识符的映射文件(制表符分隔,gene_id<制表符>trans_id<回车>)
-m <int> 最小蛋白质长度(默认值:100)
-S 特定链(仅分析顶链)
--output_dir | -O <string> 意图输出目录的路径
--version 显示版本标签(5.7.1)
--genetic_code | -G <string> 遗传密码(默认值:普遍;见 PerlDoc;选项:Euplotes, Tetrahymena, Candida, Acetabularia) 遗传密码(来源于:https://www.ncbi.nlm.nih.gov/Taxonomy/Utils/wprintgc.cgi)
Acetabularia;Candida;纤毛虫;Dasycladaceae;Euplotid;Hexamita;Mesodinium;线粒体-海鞘;
线粒体-绿藻;线粒体-棘皮动物;线粒体-扁形动物;线粒体-无脊椎动物;线粒体-原生动物;线粒体-Pterobranchia;线粒体-Scenedesmus obliquus;线粒体-Thraustochytrium;线粒体-吸虫;线粒体-脊椎动物;线粒体-酵母;Pachysolen tannophilus;Peritrich;SR1_Gracilibacteria;Tetrahymena;普遍;
--complete_orfs_only 只产出完整的 ORFs(peps 以 Met (M) 开始,以 stop (*) 结束)
#2 第二步
./TransDecoder.Predict -h
Transdecoder.LongOrfs - 转录组蛋白质预测
必需项:
-t <string> 文件名为 transcripts.fasta 的转录本
常用选项:
--retain_long_orfs_mode <string> 'dynamic' 或 'strict'(默认:dynamic) 在动态模式下,根据同 GC 含量的随机序列中的 1%FDR 设置范围。
--retain_long_orfs_length <int> 在 'strict' 模式下,保留所有找到的等于或长于这么多核苷酸的 ORF,即使没有其他证据将其标记为编码(默认:1000000),因此默认情况下基本上是关闭的。
--retain_pfam_hits <string> 从运行 hmmscan 搜索 Pfam 而得到的域表输出文件(详见 transdecoder.github.io) 任何具有 pfam 域命中的 ORF 将被保留在最终输出中。
--retain_blastp_hits <string> 以 '-outfmt 6' 格式的 blastp 输出。 任何具有 blast 匹配的 ORF 将被保留在最终输出中。
--single_best_only 仅保留每个转录本的单个最佳 ORF(优先考虑同源性然后是 ORF 长度)
--output_dir | -O <string> 与 TransDecoder.LongOrfs 相同的输出目录
--no_refine_starts 开始细化使用 PWM 识别 5' 部分 ORF 的潜在起始密码子,默认情况下开启此过程。
高级选项
-T <int> 用于训练马尔可夫模型(六聚体统计)的最长 ORF 数量(默认:500) 注意,首先选择 10 倍的这个值来去除冗余,然后从非冗余集中选择这个 -T 值的最长 ORF。
--version 显示版本(5.7.1)
--genetic_code | -G <string> 遗传密码(默认:普遍;见 PerlDoc;选项:Euplotes, Tetrahymena, Candida, Acetabularia, ...) 遗传密码(来源于:https://www.ncbi.nlm.nih.gov/Taxonomy/Utils/wprintgc.cgi)
Acetabularia;Candida;纤毛虫;Dasycladaceae;Euplotid;Hexamita;Mesodinium;线粒体-海鞘;
线粒体-绿藻;线粒体-棘皮动物;线粒体-扁形动物;线粒体-无脊椎动物;线粒体-原生动物;线粒体-Pterobranchia;线粒体-Scenedesmus obliquus;线粒体-Thraustochytrium;线粒体-吸虫;线粒体-脊椎动物;线粒体-酵母;Pachysolen tannophilus;Peritrich;SR1_Gracilibacteria;Tetrahymena;普遍;4.1 官方protocol
#运行 TransDecoder 注意,TransDecoder 现在可以在 Galaxy 工具上使用
从转录本 fasta 文件预测编码区域
“TransDecoder”工具在包含目标转录本序列的 fasta 文件上运行。最简单的用法如下:
步骤 1:提取长的开放阅读框
TransDecoder.LongOrfs -t target_transcripts.fasta
默认情况下,TransDecoder.LongOrfs 将识别至少有 100 个氨基酸长的 ORFs。您可以通过 '-m' 参数降低这个长度阈值,但请知道,随着最小长度标准的缩短,错误阳性 ORF 预测的比率会急剧增加。
如果转录本按照正义链定向,则包括 -S 标志以仅检查顶链。完整的使用信息如下。
如果你想排除部分 ORF,请包含 --complete_orfs_only,但请注意,在 5' 部分的情况下,所选的起始密码子可能不正确,因为 ORF 向上游延伸。同样,3' 部分和完全读通(没有起始和没有停止)的转录本序列将被排除。
步骤 2:(可选)
可选地,通过 blast 或 pfam 搜索识别与已知蛋白质同源的 ORFs。
见下方的包含同源性搜索作为 ORF 保留标准的部分。
步骤 3:预测可能的编码区域
TransDecoder.Predict -t target_transcripts.fasta [ 同源性选项 ]
最终的候选编码区域集可以在扩展名包括 .pep、.cds、.gff3 和 .bed 的文件 '.transdecoder.' 中找到
从基于基因组的转录本结构 GTF 文件开始(例如,cufflinks 或 stringtie)
这里的过程与上述相同,不同之处在于我们必须首先生成对应于转录本序列的 fasta 文件,并且最后,我们重新计算描述基因组背景下预测编码区域的 GFF3 格式的基因组注释文件。
使用基因组和转录本.gtf 文件构建转录本 fasta 文件,如下所示:
util/gtf_genome_to_cdna_fasta.pl transcripts.gtf test.genome.fasta > transcripts.fasta
接下来,将转录本结构 GTF 文件转换为 alignment-GFF3 格式的文件(这样做只是因为我们的过程是基于 gff3 而不是起始的 gtf 文件操作的 - 没有什么大不了的)。使用 cufflinks GTF 输出作为示例,将 gtf 转换为 alignment-gff3,如下所示:
util/gtf_to_alignment_gff3.pl transcripts.gtf > transcripts.gff3
现在,按照上述过程运行以生成您最佳的候选 ORF 预测:
TransDecoder.LongOrfs -t transcripts.fasta
(可选地,识别与已知蛋白质同源的肽段)
TransDecoder.Predict -t transcripts.fasta [ 同源性选项 ]
最后,生成一个基于基因组的编码区域注释文件:
util/cdna_alignment_orf_to_genome_orf.pl \
transcripts.fasta.transdecoder.gff3 \
transcripts.gff3 \
transcripts.fasta > transcripts.fasta.transdecoder.genome.gff34.2如何与BLAST等连用
blast使用详见Blast安装及使用-Blast+2.14.0(bioinfomatics tools-001)
如何确定自己得到的unigene为真实基因,结合数据库检索是一个好的方法,使用swiss或者Pfam数据库进行初步匹配,提高序列的真实性!这也为后续qPCR或者PCR能出现条带奠定基础。
将同源性搜索包含为 ORF 保留标准
为了进一步提高捕获可能具有功能意义的 ORFs 的灵敏度,不论上述的编码可能性得分如何,您可以扫描所有 ORFs 以寻找与已知蛋白质的同源性,并保留所有这类 ORFs。这可以通过两种流行的方式完成:针对已知蛋白质数据库进行 BLAST 搜索,以及搜索 PFAM 以识别常见的蛋白质结构域。在 TransDecoder 的上下文中,这是这样完成的:
运行 TransDecoder.LongOrfs 后,您会找到一个名为 '${transcripts_file}.transdecoder_dir/longest_orfs.pep' 的多 fasta 蛋白质文件。使用以下方法搜索这些候选肽段的同源性:
BlastP 搜索
使用 BLAST+ 搜索如 Swissprot(快速)或 Uniref90(慢但更全面)这样的蛋白质数据库
一个示例命令可能如下所示:
```bash
blastp -query transdecoder_dir/longest_orfs.pep \
-db uniprot_sprot.fasta -max_target_seqs 1 \
-outfmt 6 -evalue 1e-5 -num_threads 10 > blastp.outfmt6
```
如果您可以使用计算网格,考虑使用 HPC GridRunner 进行更高效的并行计算。
有些人更喜欢使用 diamondblast 而不是 NCBI blast 以大大提高速度 - 但要注意潜在的灵敏度差异。
Pfam 搜索
使用 Pfam 搜索肽段的蛋白质结构域。这需要安装 hmmer3 和 Pfam 数据库。
```bash
hmmsearch --cpu 8 -E 1e-10 --domtblout pfam.domtblout /path/to/Pfam-A.hmm transdecoder_dir/longest_orfs.pep
```
这里应该可以使用 hmmscan 和 hmmsearch,但由于执行速度更快,建议使用 hmmsearch。
就像 blast 搜索一样,如果您可以使用计算网格,考虑使用 HPC GridRunner。
将 Blast 和 Pfam 搜索结果整合到编码区域选择中
上述生成的输出可以被 TransDecoder 利用,以确保那些具有 blast 命中或域命中的肽段被保留在报告的可能编码区域集合中。像这样运行 TransDecoder.Predict:
```bash
TransDecoder.Predict -t target_transcripts.fasta --retain_pfam_hits pfam.domtblout --retain_blastp_hits blastp.outfmt6
```
最终的编码区域预测现在将包括那些具有与编码区域一致的序列特征以及那些显示出 blast 同源性或 pfam 结构域内容的区域。4.3 输出结果解读
输出文件解释
一个工作目录(例如 transcripts.transdecoder_dir/)被创建来运行和存储管道的中间部分,并包含:
longest_orfs.pep:满足最小长度标准的所有 ORFs,不论其编码潜力如何。
longest_orfs.gff3:在目标转录本中找到的所有 ORFs 的位置
longest_orfs.cds:所有检测到的 ORFs 的核苷酸编码序列
longest_orfs.cds.top_500_longest:用于训练编码序列的马尔可夫模型的前 500 个最长 ORFs。
hexamer.scores:每个 k-mer 的对数似然分数(编码/随机)
longest_orfs.cds.scores:每个 ORF 在 6 个阅读框架中的对数似然总分数
longest_orfs.cds.scores.selected:基于评分标准(顶部描述)选择的 ORFs 的存取号
longest_orfs.cds.best_candidates.gff3:转录本中选定 ORFs 的位置
然后,最终输出在您当前的工作目录中报告:
transcripts.fasta.transdecoder.pep:最终候选 ORFs 的肽序列;所有较短的候选项都从较长的 ORFs 中移除。
transcripts.fasta.transdecoder.cds:最终候选 ORFs 的编码区域的核苷酸序列
transcripts.fasta.transdecoder.gff3:最终选定 ORFs 在目标转录本中的位置
transcripts.fasta.transdecoder.bed:描述 ORF 位置的 bed 格式文件,最适合使用 GenomeView 或 IGV 查看。
java -jar $GENOMEVIEW/genomeview.jar transcripts.fasta transcripts.fasta.transdecoder.bed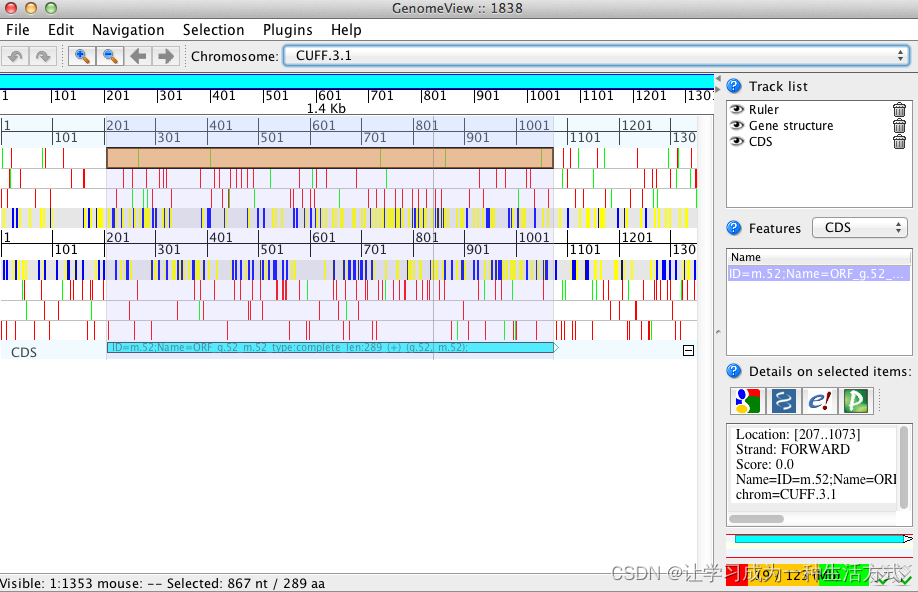
java -jar $GENOMEVIEW/genomeview.jar test.genome.fasta transcripts.bed transcripts.fasta.transdecoder.genome.bed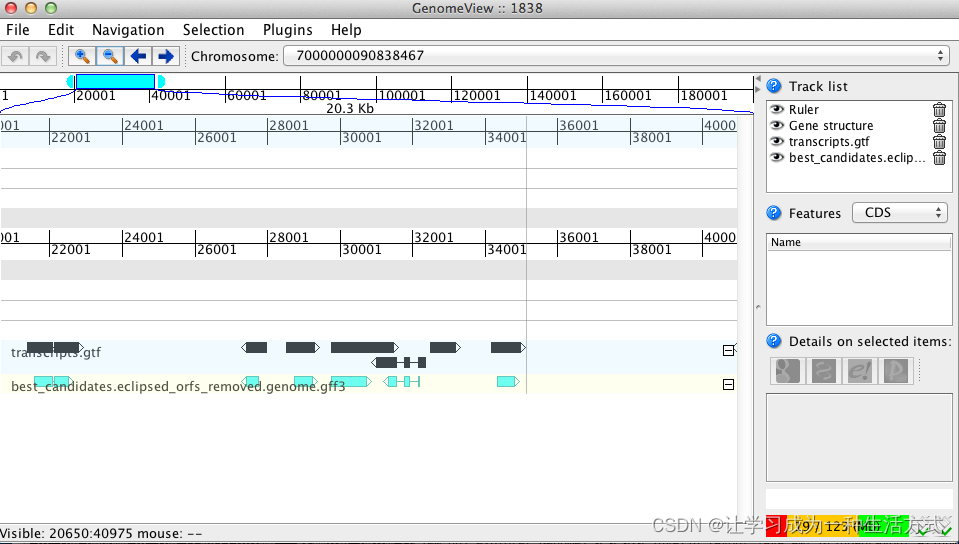
这里使用两个软件GenomeView or IGV 实现了预测CDS结构的可视化,当然Apollo等软件也可做到这一点。
05 常用命令行
TransDecoder 去冗余三部曲,这里是和配合trinity使用,详见Trinity安装与使用-Trinity-v2.15.1(bioinfomatics tools-006)
##预测三部曲
./TransDecoder-v5.7.1/TransDecoder.LongOrfs -t Trinity.fasta
./blastp -d ./uniprot_sprot.fa.db.dmnd -q ./longest_orfs.pep --evalue 1e-5 --max-target-seqs 1 > XXblastp.outfmt6
/new2pan/xyy230105/230817p450jinhuawenxianjishuluxian/001p450apps/TransDecoder-v5.7.1/TransDecoder.Predict -t ./Trinity.fasta --retain_blastp_hits XXblastp.outfmt6 06 参考文献
李静. 无花果果皮花青苷积累的转录组分析及调控机理研究[D]. 南京农业大学, 2020. DOI:10.27244/d.cnki.gnjnu.2020.000274.
朱峰林. 莲基因组重测序、转录组分析及淀粉合成相关基因克隆[D]. 武汉大学, 2019. DOI:10.27379/d.cnki.gwhdu.2019.000094.
Amombo E . 高羊茅和狗牙根的连锁不平衡,转录组分析和耐盐激素调节[D]. 中国科学院大学(中国科学院武汉植物园), 2019. DOI:10.27603/d.cnki.gkhzs.2019.000024.

















 7871
7871

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









