









学习记录,以下载《Functional traits and adaptation of lake microbiomes on the Tibetan Plateau》中的宏基因组为例。
1、找到SRP项目号
1.1、找到文章Data availablity中的NCBI编号

这里要说明一下这些编号的含义:
在NCBI、ENA(欧洲核酸档案馆)和DDBJ(日本DNA数据库)中,PRJNA、PRJEB和PRJDB是用于标识生物测序研究项目(BioProject)的编号前缀,属于国际核苷酸序列数据库联盟(INSDC)的标准命名规则。
PRJNA
- 所属数据库:NCBI BioProject(美国国家生物技术信息中心)。
- 含义:以
PRJNA开头的编号(如PRJNA725666)是NCBI BioProject数据库中的唯一项目标识符。- 用途:
- 标识一个完整的测序研究项目(例如基因组测序、转录组分析、宏基因组研究)。
- 关联该项目的所有子数据(如样本、实验、原始测序数据)。
- 示例:
PRJNA725666:新冠病毒基因组监测项目。- 包含子数据:SRA测序数据(如
SRR14445601)、样本信息(如SAMN18927417)。
SRA登录号(Sequence Read Archive)
- 含义:标识高通量测序原始数据,格式为
SRR/SRX/SRS+数字(如SRR1234567)。- 层级关系:
- SRP:研究项目(Study)。
- SRS:样本(Sample)。
- SRX:实验(Experiment)。
- SRR:测序运行数据(Run)。
- 用途:下载原始测序数据(如RNA-Seq、ChIP-Seq)。
1.2、打开NCBI官网
National Center for Biotechnology Information (nih.gov)
直接输入NCBI的编号
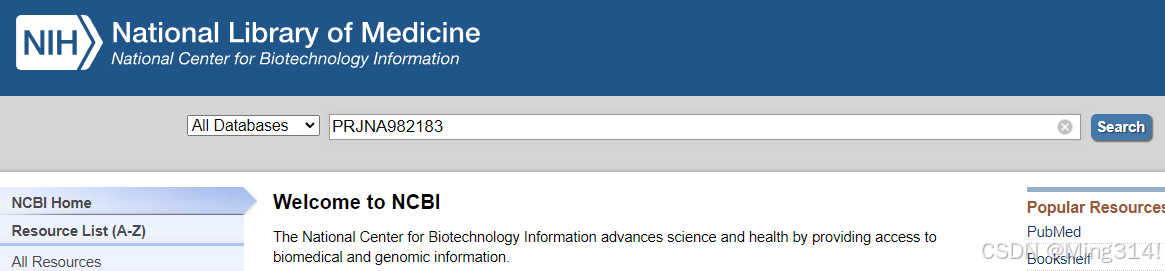
搜索后出现bioproject
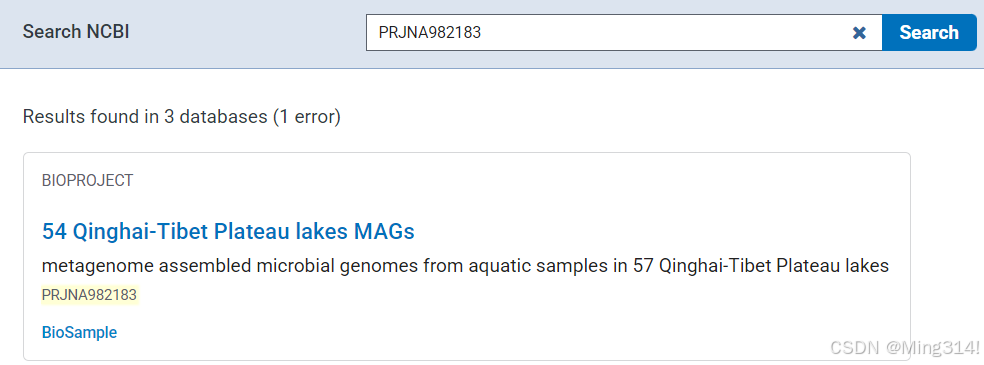
根据上面编号的含义,我们应该下拉找SRA
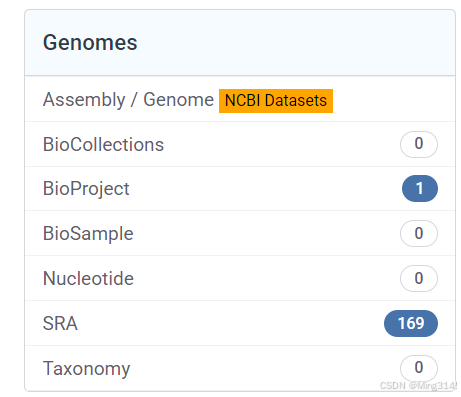
这个里面就是这篇文章包含的所有数据,我们选择其中一个打开

这里面就有它的基本信息,可以看到有SRP443144,这个就是研究项目的编号。

2、进入NCBI SRA数据库搜索项目界面,输入SRP编号
https://www.ncbi.nlm.nih.gov/Traces/study/
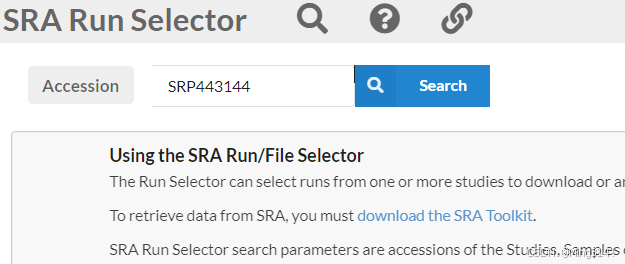
进入后,下面这个 Found 169 items,就是测序数据。
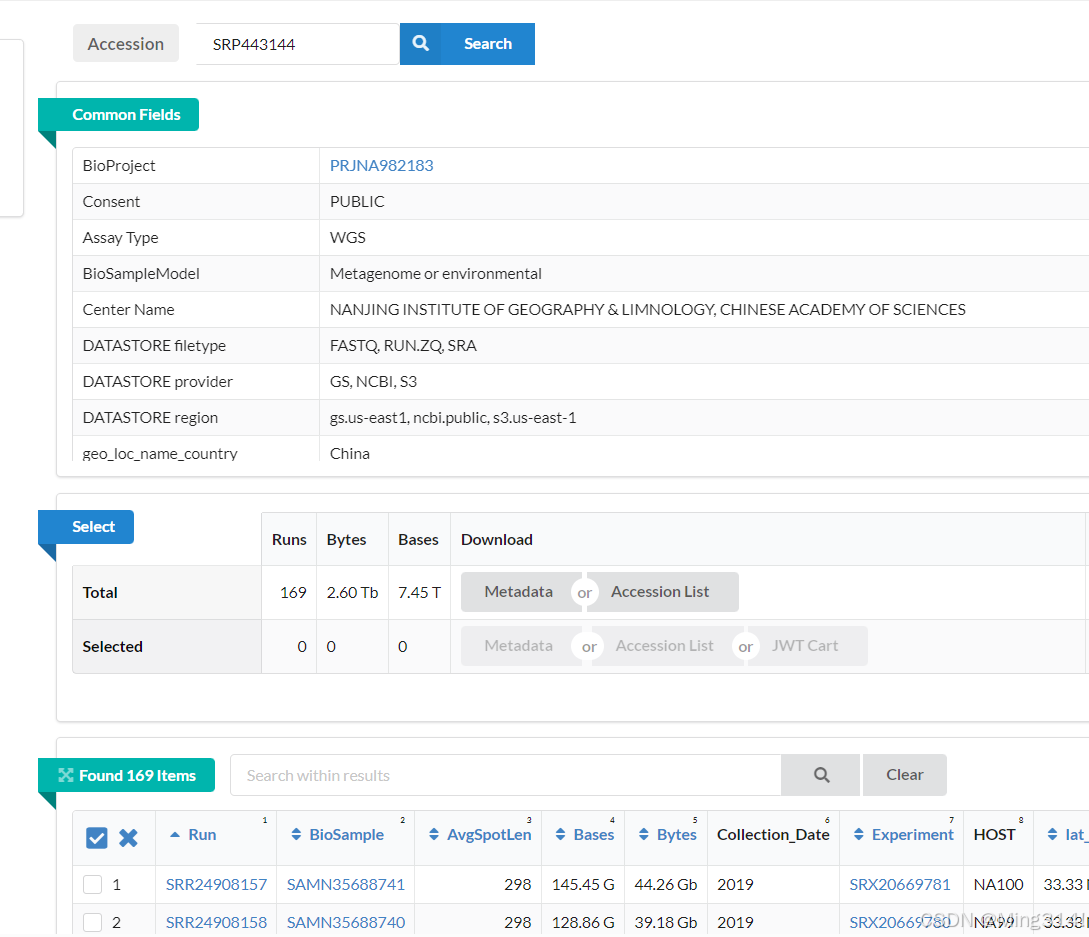
我们选择我们需要的数据,然后在select部分选择Selected的Accession list 下载下来。
3、下载
3.1 SRA Toolkit工具下载
下载地址:https://github.com/ncbi/sra-tools/wiki/01.-Downloading-SRA-Toolkit
这个里面有很多可以选择,我选择的是windows系统的,我个人觉得方便一些,习惯使用linux系统的同学也可以下载linux版本。

windows下载之后就是一个zip的压缩包,解压之后就可以直接使用了。
3.2 批量下载
将Accession list放在bin的文件夹下面
![]()
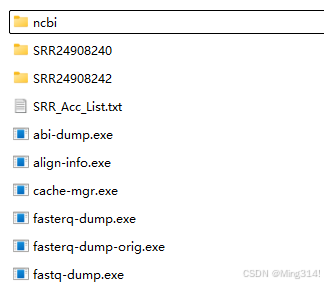
然后使用NCBI的SRA Toolkit进行批量的下载,程序会逐个下载各个样本的数据,一个样本一个文件夹,每个文件夹里面都是一个sra文件
prefetch.exe --option-file SRR_Acc_List.txt得到的sra文件不能直接使用,要转换成fastq文件
fastq-dump -I --split-files SRR1234567.sraSRR1234567.sra是下载的数据文件名及后缀。 得到fastq双端测序数据后就可以进行后面的分析啦
小Tips:
1、终端如何调出
我们已经选择windows系统的SRA Toolkit了,就说明我们对于这些指令可能并不熟悉,所以这条是写给不太熟悉代码操作的同学的,大佬可以跳过哈
因为这个还需要写明你的路径(如下图),所以最直接的方式就是在这个文件夹的空白地方右击,就会显示一个在终端中打开,选择后,打开的终端就是你的操作路径了,简单直接。
![]()
2、代码报错
可以在上图中看的,我的代码前面加了一个.\ 这个也是我在运行中出现的问题,明明可以找到文件但还是不能运行,加一个.\ 就可以了。
3、转换成fastq时报错
我们下载sra文件时,是一个样本一个文件夹,每个文件夹里面都是一个sra文件,所以我们下意识的可能会在sra的文件夹里单独操作,这个时候就会报错,是因为:我们是依赖bin下面的这些exe进行操作的,所以sra文件夹中并没有这个exe,我们只需要把exe结尾的都复制进去,再输入代码,就可以正常操作啦~
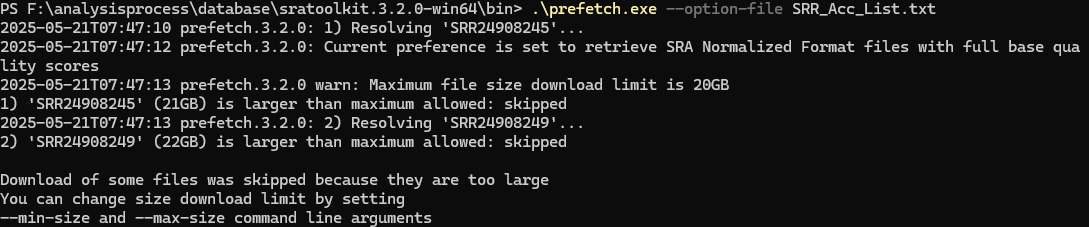
4、文件过大不能下载
我们下载的时候可能会出现一下报错,是因为默认的下载文件大小是20GB,超过这个大小,就需要通过 --max-size来设置,我一般会直接设置成100GB,然后最重要的是,它是以KB为单位的,所以100GB,要写成:--max-size 104857600


















 1285
1285

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











