






α多样性主要关注局域均匀生境下的物种数目,因此也被称为生境内的多样性(within-habitatdiversity)。群落生态学中研究微生物多样性,通过单样品的多样性分析(α多样性)可以反映微生物群落的丰度和多样性,包括一系列统计学分析指数估计环境群落的物种丰度和多样性。
α分类有多种,分别表达物种的丰富度、均匀度及多样性等。
多样性指数(Community Diversity)反映的是物种丰富度和均匀度的综合状况,常见的有Shannon、Simpson等。Shannon指数反映的是物种丰度与均匀度,与这两者均呈正相关;Simpson指数为在样本中抽取两条序列属于不同种的概率。Shannon指数越高表明α多样性越高,而Simpson相反(但当前很多Simpson是1-x)。
丰富度指数(Community Richness)反映的是群落内物种的丰富程度,常见的有Sobs、Chao、ACE等。Sobs为该样本实际包含的OTU(或ASV等)的物种数目。而Chao为修正后的物种数目。(有的文章中Sobs也称Richness)
接下来上代码!(该处使用宏基因组数据rpkm作为丰度表,有小数)
#先加载可能需要的所有包。
library(vegan)
#加载数据
df = read.delim("C:/Users/Shizhe Zhang/Desktop/VOTU.txt", row.names=1,header = T, sep = "\t")
#可以查看用法
#?diversity()
Shannon = diversity(df, index = "shannon", MARGIN = 2, base = exp(1)) #MARGIN决定横竖,base为底,此处为e
Simpson = diversity(df, index = "simpson", MARGIN = 2, base = exp(1))
Richness = specnumber(df, MARGIN = 2) # Sobs
result1$pielou = Shannon/log(Richness,2)
#以数字形式统计成表格并合并
result1 = as.data.frame(cbind(Shannon, Simpson, Richness))
#加载分组文件
group = read.delim("C:/Users/HUAWEI/Desktop/group.txt", header = T)
result1$group=group[,2] # 合并分组文件
shannon= ggplot(data=result1, aes(y=Shannon, fill=group,x=group)) + #确定xy和颜色
stat_boxplot(geom = "errorbar",width = 0.3)+ #添加误差棒
geom_boxplot() + #画箱线图
theme_classic()+ #取消背景
labs(y = "Shannon",x= "Group") #标注xy轴名字df格式如下:
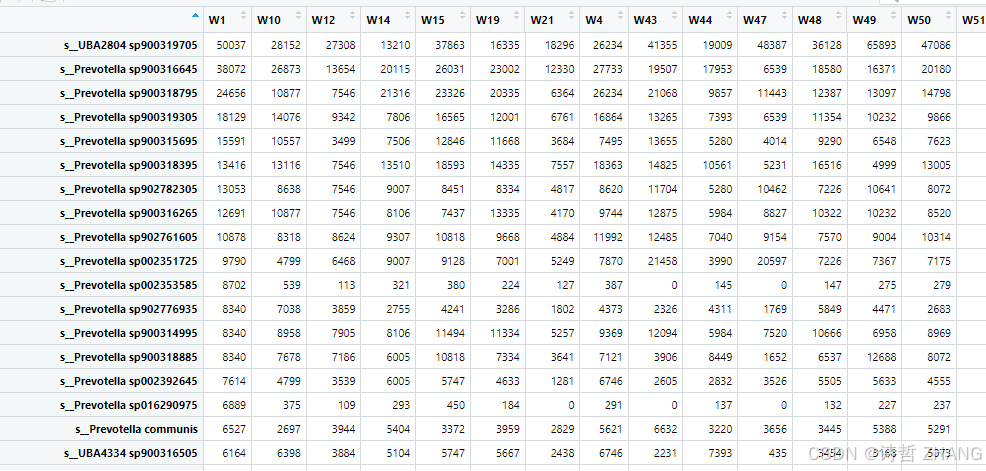 group格式如下:
group格式如下:
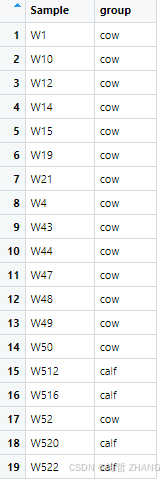

















 1354
1354

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









