







本文背景资料(感谢Jimmy大神):RNA-seq基础入门传送门-转录组-生信技能树 (biotrainee.com)
fastq-dump参数参考文章(有些已经过时啦!!):SRA批量下载及转为Fastq格式 - 简书 (jianshu.com)
相关软件的下载地址:
sratoolkit:https://ftp-trace.ncbi.nlm.nih.gov/sra/sdk/2.11.3/sratoolkit.2.11.3-ubuntu64.tar.gz
fastqc: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/fastqc_v0.11.9.zip
hisat2: https://cloud.biohpc.swmed.edu/index.php/s/oTtGWbWjaxsQ2Ho/download
目录
一、数据转换
需要转换的sra文件:
for i in `seq 56 62`;do nohup fastq-dump --gzip --split-files -O ../raw_data/ SRR35899$i.sra; done &/*
seq 56 62生成一个56到62的列表,` `符号取值。
nohup指出即使关闭终端,程序仍会运行;末尾的&符号,让程序在后台运行;可以使用jobs -l显示所有在后台运行的程序,kill 进程号关闭某个进程。
fastq-dump 用来将sra转变为fastq文件;--gzip指定输出文件为压缩格式;--split-files将reads分开到两个文件,文件末尾会有后缀;-O指定输出文件夹;
*/
结果:
二、fastqc检测测序文件质量
注意一下:我这fastqc好像不支持压缩文件(gz)的输入?倒腾了十分钟,最后把压缩文件给解压缩了才能运行
#切换fastq所在文件目录
ls | xargs nohup fastqc -o ../fastqc_result/ --nogroup &
xargs和管道符|的区别(抄的):[Linux] xargs 和 管道符的区别 - 我是小菜鸟 - 博客园 (cnblogs.com)
xargs相当于传给后面一个参数,而管道则传给后面命令一个字符串
上述代码将目录下的所有fastq文件依次传给后面的fastqc进行检测,结果输出到../fastqc_result文件夹中。
部分结果(Html+zip):
fastqc结果解释:用FastQC检查二代测序原始数据的质量 | Public Library of Bioinformatics (plob.org)
可以使用multiqc将结果结合在一起,更加直观:批量显示QC结果的利器 | 分享自为知笔记 (wiz03.com)
multiqc -o ../multiqc_result/ ../fastqc_result/
结果:

将这个html文件下载到本地,使用浏览器打开即可。
三、下载基因组参考文件、建序和注释文件
去UCSC下载hg19参考基因组:http://genome.ucsc.edu/index.html
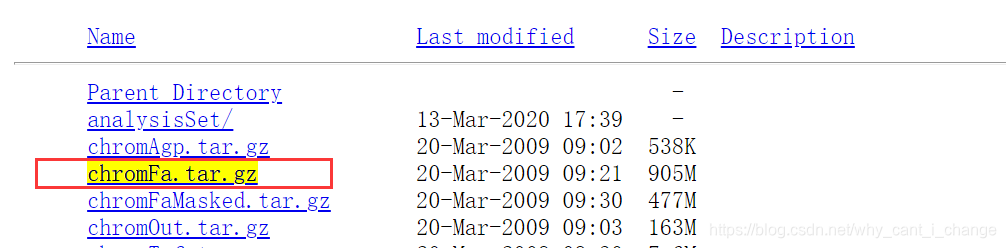
chromFa.tar.gz - The assembly sequence in one file per chromosome.
Repeats from RepeatMasker and Tandem Repeats Finder (with period
of 12 or less) are shown in lower case; non-repeating sequence is
shown in upper case.
再去GENCODE - Home page下载注释文件,参考文章(伪)从零开始学转录组:了解参考基因组及基因注释 (qq.com)
GTF等格式参考官网:GENCODE - Data format (gencodegenes.org)
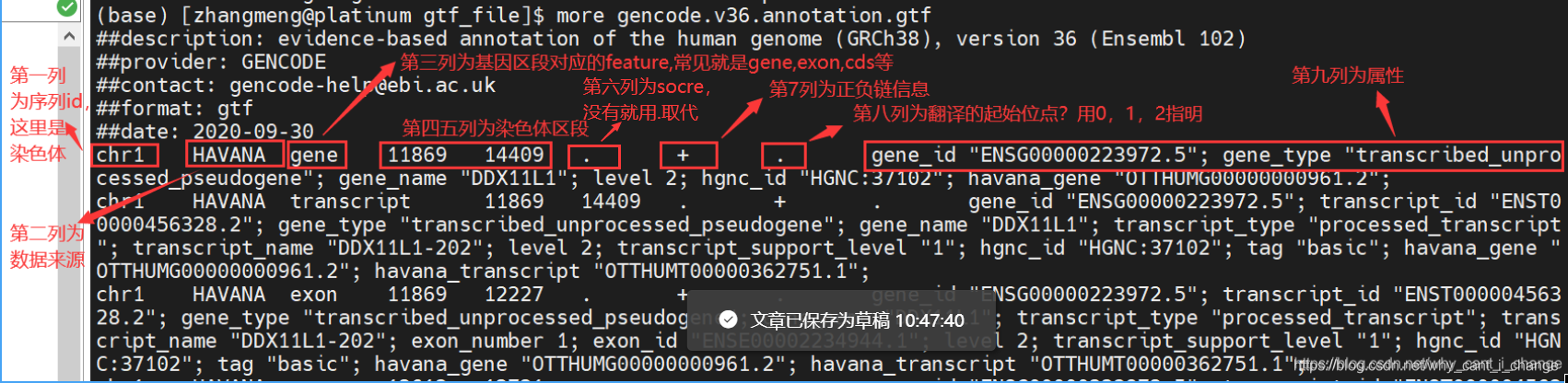
关于为什么还要指明正负链,参考文章:关于DNA正负链的定义 - 简书
安装IGV(Integrative Genomics Viewer),也就是基因组浏览器,之前已经安装好了。
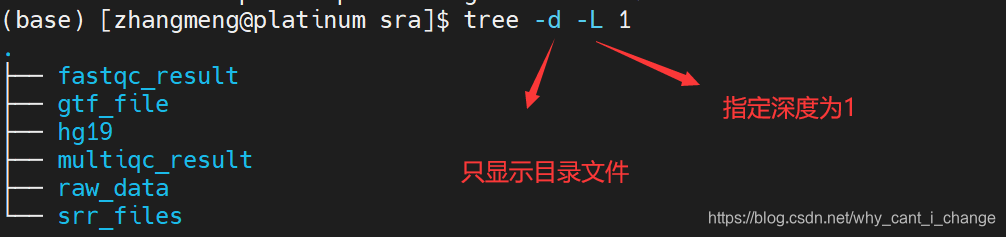
至此,转录组分析所需要的材料就全了,目前的结构目录如下:
如果之前使用过bowtie或者tophat的童鞋都知道,下一步就要给reference genome建序了,这里我们可以去hisat2官网上直接下载hg19已经建好的index:Download | HISAT2 (daehwankimlab.github.io)
四、使用hisat2进行比对
Usage:
hisat2 [options]* -x <ht2-idx> {-1 <m1> -2 <m2> | -U <r> | --sra-acc <SRA accession number>} [-S <sam>]
<ht2-idx> Index filename prefix (minus trailing .X.ht2).
<m1> Files with #1 mates, paired with files in <m2>.
Could be gzip'ed (extension: .gz) or bzip2'ed (extension: .bz2).
<m2> Files with #2 mates, paired with files in <m1>.
Could be gzip'ed (extension: .gz) or bzip2'ed (extension: .bz2).
<r> Files with unpaired reads.
Could be gzip'ed (extension: .gz) or bzip2'ed (extension: .bz2).
<SRA accession number> Comma-separated list of SRA accession numbers, e.g. --sra-acc SRR353653,SRR353654.
<sam> File for SAM output (default: stdout)
<m1>, <m2>, <r> can be comma-separated lists (no whitespace) and can be
specified many times. E.g. '-U file1.fq,file2.fq -U file3.fq'.
那么在本例中,可以使用一个for循环来执行:
for i in `56 58`
do
nohup hisat2 -x reference/index/hg19_genome -1 raw_data/SRR35899$i_1.fastq -2 raw_data/SRR35899$i_2.fastq -S aligned/SRR35899$1.sam &
done结果:
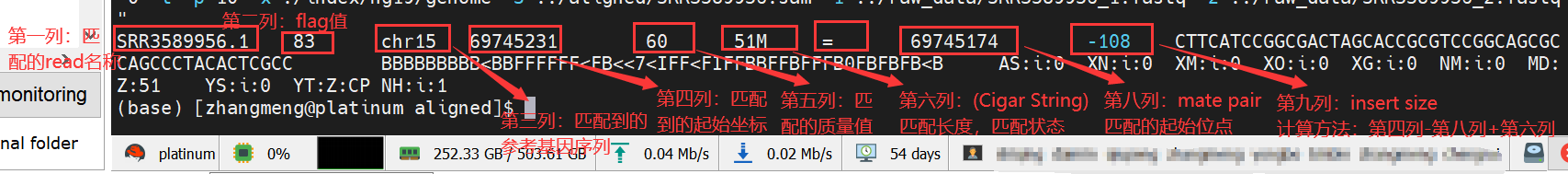
sam格式,参考生信:2:sam格式文件解读_genome_denovo的博客-CSDN博客_sam文件
五、samtools进行处理
参考文章:BAM/SAM文件操作与可视化简介
SAM(sequence Alignment/mapping)数据格式是目前高通量测序中存放比对数据的标准格式;而目前处理SAM格式的工具主要是SAMTools:
-
view: BAM-SAM/SAM-BAM 转换和提取部分比对
-
sort: 比对排序
-
index: 索引排序比对
最常用的三个命令就是格式转换,排序,索引:
格式转换当然就是为了省空间提速度啦,这咱们也不懂,用就完事了~
#转换格式sam->bam,压缩空间同时利于下面处理
#-b指明输出为bam格式,S指明自动检测输入的格式,-o指明输出文件名
$ for i in `seq 56 58`
$ do
$ nohup samtools view -bS -o SRR35899${i}.bam SRR35899${i}.sam &
$ done
那为啥要排序呢(sort),你看看上面的sam文件格式,比对是每一条read比对到参考基因组上,那么就有可能第一条read比对到chr10,第二条却比对到了chr2,所以要调一调顺序哈~
$ for i in `seq 56 58`
$ do
$ nohup samtools sort --threads 50 SRR35899${i}.bam -o SRR35899${i}_sorted.bam &
$ done最后对排序好的bam文件建立索引,加快后面检索速度。
$ for i in `seq 56 58`
$ do
$ nohup samtools index -@ 50 SRR35899${i}_sorted.bam &
$ done结果: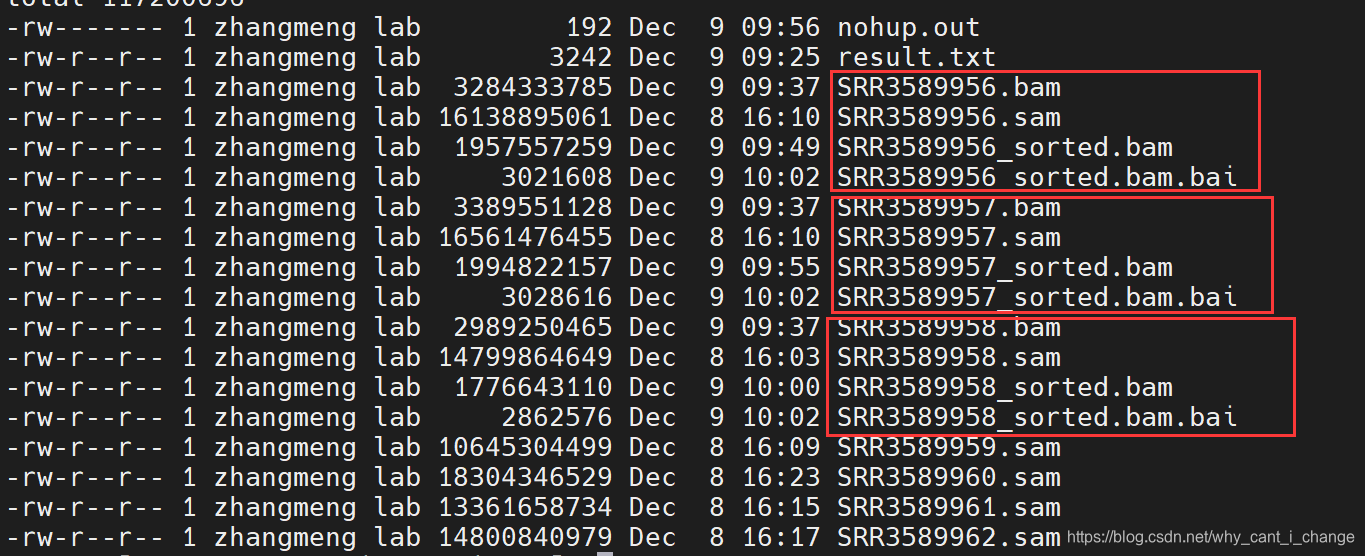
至于为什么只用56-58的,参考文章里提到了:(伪)从零开始学转录组(5) 序列比对
同时如果你自己用samtools flagstat去看一下比对的情况,你就会发现后面几个结果差的离谱。

















 4496
4496

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









