



 本文详细介绍FASTA和FASTQ两种生物学数据文件格式的特点及应用。FASTA用于存储已知序列,而FASTQ则用于存储测序原始数据,两者均为纯文本格式。文章深入解析了这两种格式的具体内容、质量值体系及其在生物信息学分析中的作用。
本文详细介绍FASTA和FASTQ两种生物学数据文件格式的特点及应用。FASTA用于存储已知序列,而FASTQ则用于存储测序原始数据,两者均为纯文本格式。文章深入解析了这两种格式的具体内容、质量值体系及其在生物信息学分析中的作用。
本文转载自微信公众号解螺旋的矿工,作者为黄树嘉,已获得授权。黄树嘉写了WGS系列的文章,堪称教科书级别的生物信息学习材料。虽然本平台只关注宏基因组领域,但此系列文章知识体系完善、干货满满,是值得每位专门从事二代测序数据分析人员必读材料。欢迎大家关注公众号解螺旋的矿工。
学习全基因组测序数据分析系列
在WGS数据的分析过程中,我们会接触到许多生物信息学/基因组学领域所特有的数据文件和它们特殊的格式,在这一节中将要介绍的FASTA和FASTQ便是其中之一二。这是我们存储核苷酸序列信息(就是DNA序列)或者蛋白质序列信息最常使用的两种文本文件,虽然看起来名字有些古怪,但它们完全是纯文本文件(如同.txt)!名字的发音分别是fast-A和fast-Q。这一篇文章内容虽然比较简单,但还是比较长,我在这里详细介绍了这两类文件的格式特点和一些在分析的时候需要考虑的地方。
FASTA
我相信许多人(包括生物信息工程师们)一定不知道FASTA这个文件的来源,竟然是一款名叫“FASTA”的比对软件!名字中最后一个字母A,其实就是Alignment的意思!但这已经是上个世纪的事情了,最初是由William. R. Pearson 和 David. J. Lipman在1988年所编写,目的是用于生物序列数据的处理。
自那之后,生物学家和遗传学家们也没做过多的考虑,就草率地决定(其实类似的‘草率’行为在组学领域经常碰到)把FASTA作为这种存储有顺序的序列数据的文件后缀【注】,这包括我们常用的参考基因组序列、蛋白质序列、编码DNA序列(coding DNA sequence,简称CDS)、转录本序列等文件都是如此,文件后缀除了.fasta之外,也常用.fa或者.fa.gz(gz压缩)。
【注】这里的序列、序列数据,指的其实就是表示DNA或者蛋白质的一条字符串。
这里再特别强调三个字:有!顺!序!说的是从1开始一个个按顺序往下排列的意思——这不也正是序列这个词的含义!

因此,我们可以通过数个数,就知道某个DNA碱基在某个基因组上的准确位置,这个位置会用所在序列的名字和所在位置来表达,比如基因数据比对的结果(下一篇会介绍),方便后续数据分析。
FASTA文件主要由两个部分构成:序列头信息(有时包括一些其它的描述信息)和具体的序列数据。头信息独占一行,以大于号(>)开头作为识别标记,其中除了记录该条序列的名字之外,有时候还会接上其它的信息。紧接的下一行是具体的序列内容,直到另一行碰到另一个大于号(>)开头的新序列或者文件末尾。下面给出一个FASTA文件的例子,这是我们人类一个名为EGFR基因的部分序列。
>ENSMUSG00000020122|ENSMUST00000138518
CCCTCCTATCATGCTGTCAGTGTATCTCTAAATAGCACTCTCAACCCCCGTGAACTTGGT
TATTAAAAACATGCCCAAAGTCTGGGAGCCAGGGCTGCAGGGAAATACCACAGCCTCAGT
TCATCAAAACAGTTCATTGCCCAAAATGTTCTCAGCTGCAGCTTTCATGAGGTAACTCCA
GGGCCCACCTGTTCTCTGGT
>ENSMUSG00000020122|ENSMUST00000125984
GAGTCAGGTTGAAGCTGCCCTGAACACTACAGAGAAGAGAGGCCTTGGTGTCCTGTTGTC
TCCAGAACCCCAATATGTCTTGTGAAGGGCACACAACCCCTCAAAGGGGTGTCACTTCTT
CTGATCACTTTTGTTACTGTTTACTAACTGATCCTATGAATCACTGTGTCTTCTCAGAGG
CCGTGAACCACGTCTGCAAT可以看到,FASTA其实很简单,但它往往都很大,比如人类基因组有30亿个碱基,就是30亿个字符存储在这样的一个文本文件中,就算是压缩也要占用约1GB的存储空间。
另外,有两个地方,我觉得有必要提及:
第一,除了序列内容之外,FASTA的头信息并没有被严格地限制。这个特点有时会带来很多麻烦的事情,比如有时我们会看到相同的序列被不同的人处理之后、甚至是在不同的网站上或者数据库中它们的头信息都不尽相同,比如以下的几种情况都是可能存在的。
>ENSMUSG00000020122|ENSMUST00000125984
> ENSMUSG00000020122|ENSMUST00000125984
>ENSMUSG00000020122|ENSMUST00000125984|epidermal growth factor receptor
>ENSMUSG00000020122|ENSMUST00000125984|Egfr
>ENSMUSG00000020122|ENSMUST00000125984|11|ENSFM00410000138465这对于程序处理来说,凌乱的格式显然是不合适的。因此后来在业内也慢慢地有一些不成文的规则被大家所使用,那就是,用一个空格把头信息分为两个部分:第一部分是序列名字,它和大于号(>)紧接在一起;第二部分是注释信息,这个可以没有,就看具体需要,比如下面这个序列例子,除了前面gene_00284728这个名字之外,注释信息(length=231;type=dna)给出这段序列的长度和它所属的序列类型。
>gene_00284728 length=231;type=dna
GAGAACTGATTCTGTTACCGCAGGGCATTCGGATGTGCTAAGGTAGTAATCCATTATAAGTAACATG
CGCGGAATATCCGGGAGGTCATAGTCGTAATGCATAATTATTCCCTCCCTCAGAAGGACTCCCTTGC
GAGACGCCAATACCAAAGACTTTCGTAAGCTGGAACGATTGGACGGCCCAACCGGGGGGAGTCGGCT
ATACGTCTGATTGCTACGCCTGGACTTCTCTT虽然这样的格式还不算是真正的标准,但却非常有助于我们的数据分析和处理,很多生信软件(如:BWA,samtools,bcftools,bedtools等)都是将第一个空格前面的内容认定为序列名字来进行操作的。
第二,FASTA由于是文本文件,它里面的内容是否有重复是无法自检的,在使用之前需要我们进行额外的检查。这个检查倒不用很复杂,只需检查序列名字是否有重复即可。但对于那些已经成为标准使用的参考序列来说,都有专门的团队进行维护,因此不会出现这种内容重复的情况,可以直接使用,但对于其它的一些序列来说,谨慎起见,最好进行检查。
FASTQ
这是目前存储测序数据最普遍、最公认的一个数据格式,另一个是uBam格式,但这篇文章中不打算对其进行介绍。上面所讲的FASTA文件,它所存的都是已经排列好的序列(如参考序列),FASTQ存的则是产生自测序仪的原始测序数据,它由测序的图像数据转换过来,也是文本文件,文件大小依照不同的测序量(或测序深度)而有很大差异,小的可能只有几M,大的则常常有几十G上百G,文件后缀通常都是.fastq,.fq或者.fq.gz(gz压缩),以下是它的一个例子:
@DJB775P1:248:D0MDGACXX:7:1202:12362:49613
TGCTTACTCTGCGTTGATACCACTGCTTAGATCGGAAGAGCACACGTCTGAA
+
JJJJJIIJJJJJJHIHHHGHFFFFFFCEEEEEDBD?DDDDDDBDDDABDDCA
@DJB775P1:248:D0MDGACXX:7:1202:12782:49716
CTCTGCGTTGATACCACTGCTTACTCTGCGTTGATACCACTGCTTAGATCGG
+
IIIIIIIIIIIIIIIHHHHHHFFFFFFEECCCCBCECCCCCCCCCCCCCCCC你可以看到它有着自己独特的格式:每四行成为一个独立的单元,我们称之为read。具体的格式描述如下:
第一行:以‘@’开头,是这一条read的名字,这个字符串是根据测序时的状态信息转换过来的,中间不会有空格,它是每一条read的唯一标识符,同一份FASTQ文件中不会重复出现,甚至不同的FASTQ文件里也不会有重复;
第二行:测序read的序列,由A,C,G,T和N这五种字母构成,这也是我们真正关心的DNA序列,N代表的是测序时那些无法被识别出来的碱基;
第三行:以‘+’开头,在旧版的FASTQ文件中会直接重复第一行的信息,但现在一般什么也不加(节省存储空间);
第四行:测序read的质量值,这个和第二行的碱基信息一样重要,它描述的是每个测序碱基的可靠程度,用ASCII码表示。
那么,重点说一下什么是质量值?顾名思义,碱基质量值就是能够用来定量描述碱基好坏程度的一个数值。它该如何才能恰当地描述这个结果呢?我们试想一下,如果测序测得越准确,这个碱基的质量就应该越高;反之,测得越不准确,质量值就应该越低。也就是说可以利用碱基被测错的概率来描述它的质量值,错误率越低,质量值就越高!如下图,红线代表错误率,蓝线代表质量值,这便是我们希望达到的效果:
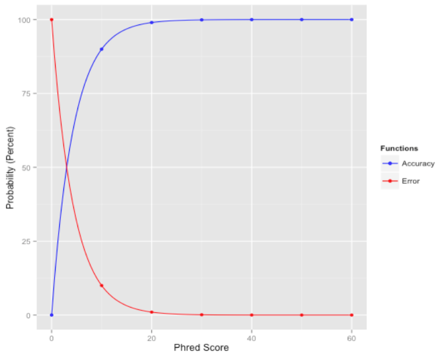
这里我们假定碱基的测序错误率为p_error,质量值为Q,它们之间的关系如下:
Q = -10log(p_error)
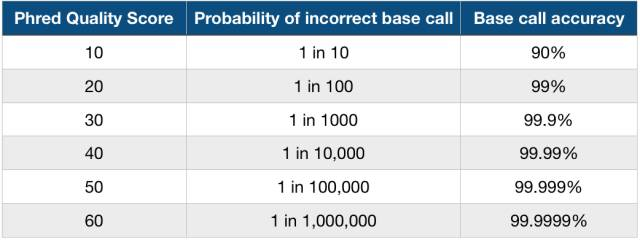
即,质量值是测序错误率的对数(10为底数)乘以-10(并取整)。这个公式也是目前测序质量值的计算公式,它非常简单,p_error的值和测序时的多个因素有关,体现为测序图像数据点的清晰程度,并由测序过程中的base calling 算法计算出来;公式右边的Q我们称之为Phred quality score,就是用它来描述测序碱基的靠谱程度。比如,如果该碱基的测序错误率是0.01,那么质量值就是20(俗称Q20),如果是0.001,那么质量值就是30(俗称Q30)。Q20和Q30的比例常常被我们用来评价某次测序结果的好坏,比例越高就越好。下面我也详细给出一个表,更进一步地解释质量值高低的含义:
现在回过头来说说为什么要用ASCII码来代表,直接用数字不行吗?行!但很难看,而且数字不能直接连起来,还得在中间加一个分隔符,长度也对不齐,还占空间,又不符合美学设计,真!麻!烦!

因此,也是为了格式存储以及处理时的方便,这个数字被直接转换成了ASCII码,并与第二行的read序列构成一一对应的关系——每一个ASCII码都和它正上面的碱基对应,这就很完美。
不过,值得一提的是,ASCII码虽然能够从小到大表示0-127的整数,但是并非所有的ASCII码都是可见的字符,比如所有小于33的ASCII码值所表示的都是不可见字符,比如空格,换行符等,因此为了能够让碱基的质量值表达出来,必须避开所有这些不可见字符。最简单的做法就是加上一个固定的整数!也的确是这么干的。
但一开始对于要加哪一个整数,并没有什么指导标准,这就导致了在刚开始的时候,不同的测序平台加的整数也不同,总的来说有以下3种质量体系,演变到现在也基本只剩下第一种了,如下表:
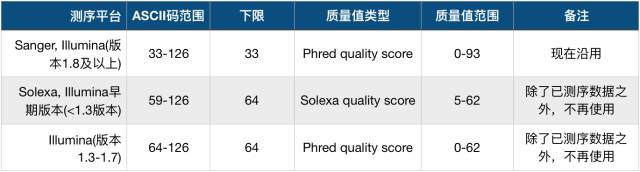
从表中可以看到下限有33和64两个值,我们把加33的的质量值体系称之为Phred33,加64的称之为Phred64(Solexa的除外,它叫Selexa64)。不过,现在一般都是使用Phred33这个体系,而且33也恰好是ASCII的第一个可见字符('!'),完美+2。
如果你在实际做项目的过程不知道所用的质量体系(经验丰富者是可以直接看出来的),那么可以用我下面这一段代码,简单地做个检查:
less $1 | head -n 1000 | awk '{if(NR%4==0) printf("%s",$0);}' \
| od -A n -t u1 -v \
| awk 'BEGIN{min=100;max=0;} \
{for(i=1;i<=NF;i++) {if($i>max) max=$i; if($i<min) min=$i;}}END \
{if(max<=126 && min<59) print "Phred33"; \
else if(max>73 && min>=64) print "Phred64"; \
else if(min>=59 && min<64 && max>73) print "Solexa64"; \
else print "Unknown score encoding"; \
print "( " min ", " max, ")";}'将上面这段代码复制到任意一份shell文件中(比如:fq_qual_type.sh),就可以用它来进行质量值类型的检查了。代码的思路其实比较简单,就是截取FASTQ文件的前1000行数据,并抽取出质量值所在的行,分别计算出其中最小和最大的ASCII值,再比较一下就判断出来了。下面给出一个例子,这是我们在本文中用到的FASTQ文件,它是Phred33的:
$ sh fq_qual_type.sh untreated.fq
Phred33
( 34, 67 )另外,在查看碱基质量值的过程中,如果你心中存有ASCII码表当然可以直接“看”出各个碱基的质量值,但在实际的场景中都是通过程序直接进行转换处理。下面我就用Python的ord()函数举个转换的例子:
In [1]: qual='JJJJJIIJJJJJJHIHHHGHFFFFFFCEEEEEDBD'
In [2]: [ord(q)-33 for q in qual]
Out[2]:
[35, 20, 17, 18, 24, 34, 35, 35, 35, 34, 35, 34, 29, 29, 32, 32, 34, 34, 33,
29, 33, 33, 32, 35, 35, 35, 34, 34, 34, 34, 35, 35, 34, 35, 34, 35, 34, 35,
34, 34, 34, 35, 35, 35, 35, 34, 33, 33, 30, 33, 24, 27]这里的ord()函数会将字符转换为ASCII对应的数字,减掉33后就得到了该碱基最后的质量值(即,Phred quality score)。
另外,根据上面phred quality score的计算公式,我们可以很方便地获得每个测序碱基的错误率,这个错误率在我们的比对和变异检测中都十分重要,后续文章中我将会讲述该部分的具体内容,以下先给出一个转换的例子,还是以上述qual为例子:
In [1]: qual='JJJJJIIJJJJJJHIHHHGHFFFFFFCEEEEEDBD'
In [2]: phred_score = [ord(q)-33 for q in qual]
In [3]: [10**(-q/10.0) for q in phred_score]
Out[3]:
[3e-04, 1e-02, 2e-02, 2e-02, 4e-03, 4e-04, 3e-04, 3e-04, 3e-04,
4e-04, 3e-04, 4e-04, 1e-03, 1e-03, 6e-04, 6e-04, 4e-04, 4e-04,
5e-04, 1e-03, 5e-04, 5e-04, 6e-04, 3e-04, 3e-04, 3e-04, 4e-04,
4e-04, 4e-04, 4e-04, 3e-04, 3e-04, 4e-04, 3e-04, 4e-04, 3e-04,
4e-04, 3e-04, 4e-04, 4e-04, 4e-04, 3e-04, 3e-04, 3e-04, 3e-04,
4e-04, 5e-04, 5e-04, 1e-03, 5e-04, 4e-03, 2e-03]这其实就是根据phred quality sxore的定义进行简单的指数运算。
小结
到这里就说完了,虽然一开始只不过是想介绍两个普通的文件格式,但写着写着就变得很长,可见,越是看似简单的东西,其实越不容易说明白。关于FASTQ还有很多需要说的内容,我打算将其留到该系列的第四篇文章里,到时我会讲述该如何构造流程对其进行有效的数据质控等,这都是构造WGS分析流程之前非常重要的内容。
我一直觉得,生物信息学(或者说基因组学)中的许多数据文件,它们的格式都有着比较特殊的一面,为了能够真正有效地进行数据分析,多花些时间搞清楚它们的细节和来龙去脉是非常重要的。不然,你有可能在后续的数据分析过程掉入意想不到的陷阱,从而浪费大量宝贵的时间去寻找可能出错的地方。

猜你喜欢
10000+:菌群分析 宝宝与猫狗 梅毒狂想曲 提DNA发Nature Cell专刊 肠道指挥大脑
文献阅读 热心肠 SemanticScholar Geenmedical
16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流、快速解决科研困难,我们建立了“宏基因组”专业讨论群,目前己有国内外1800+ 一线科研人员加入。参与讨论,获得专业解答,欢迎分享此文至朋友圈,并扫码加主编好友带你入群,务必备注“姓名-单位-研究方向-职称/年级”。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍末解决群内讨论,问题不私聊,帮助同行。

学习16S扩增子、宏基因组科研思路和分析实战,关注“宏基因组”



















 1537
1537

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









