






个人认为,生物信息学本质上是利用多种交叉学科的思想和技术对大批量数据进行处理分析、最终筛选出有用信息的一个过程。在学习生信的时候,面对同样的数据我们可能有很多方法去处理它,但这一过程往往会产生一个问题,那就是重复造轮子。比如:「现在有一个包含数百条蛋白序列的fasta文件,可你只想要序列长度小于300的蛋白,该怎么处理手头的数据?」
Python当然是一个很好的选择,但想要用python处理这样一个简单的问题,免不了十几行的代码量,对于分分钟和时间赛跑、时时刻刻被试验push的广大小镇实验家们,无疑是一种重复造轮子的巨大浪费。
Awk的存在很好的解决了这个问题,它提供了一个现成的轮子可以直接安在我们全马力的科研拖拉机上,给我们提供充足的便利,可以放心驰骋在发paper、拿offer的大路上。
言归正传,回到最初的那个问题,现在有一个包含数百条蛋白序列的fasta文件,可你只想要序列长度小于300的蛋白,该怎么处理手头的数据?
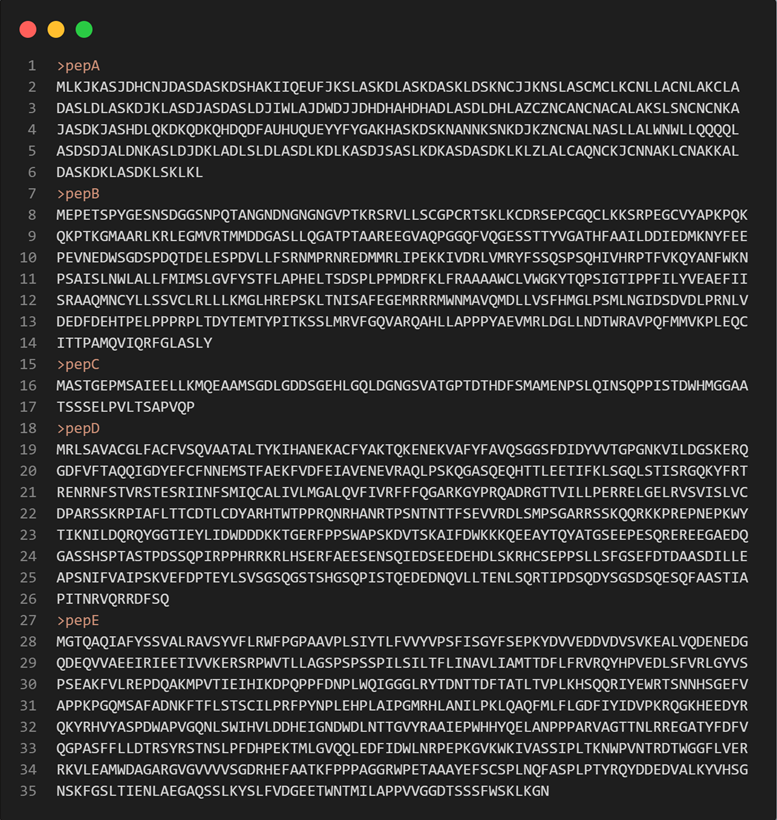
单行awk可解:
awk 'BEGIN{FS="\t";OFS="\t"}{if ($0~/^>/){id=$0;next}else{seq[id]=seq[id]$0}}END{for (j in seq) {if (length(seq[j])<300) print j"\n"seq[j]}}' test.fasta >output.fasta输出结果如下:
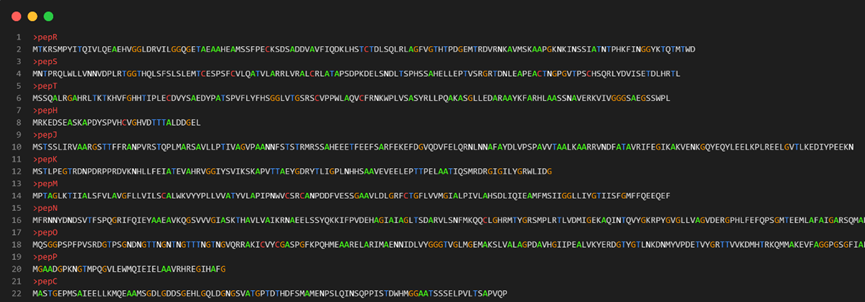
如果只想知道序列id号,只需稍加改变:
awk 'BEGIN{FS="\t";OFS="\t"}{if ($0~/^>/){id=$0;next}else{seq[id]=seq[id]$0}}END{for (j in seq) {if (length(seq[j])<300) print substr(j,2)}}' test.fasta > outputID.list输出结果如下:
想知道每一个蛋白对应的具体长度,awk也可以轻松搞定:
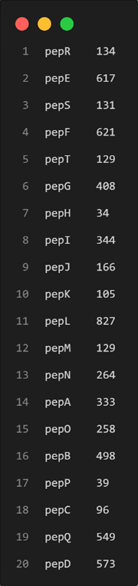
awk 'BEGIN{FS="\t";OFS="\t"}{if ($0~/^>/){id=$0;next}else{len[id]+=length($0)}}END{for (i in len){print substr(i,2),len[i]}}' test.fasta > outputLen.txt让我们在这里继续发散思维,一题多解,用另一种思路解决这个问题:
awk '/>/&&NR>1{print ""}{printf "%s",/^>/ ? $0" " : $0}' test.fasta | awk '{print$1"\t"length($2)}' > outputLen.txt*其中[condition ? command1 : command2]) 相当于if/else的简洁写法,需要注意的是:和?前后均有空格。
输出结果如下:
单行awk给了生信工作者另一种可能,在很大程度上缓解了大家的代码压力。当然,awk本质上作为一种编程语言,其深度和广度依然是值得大家深入学习的。今后,会为大家分享更多的awk使用小技巧。

















 1952
1952

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









