






使用geNomad识别移动遗传元素
Identification of mobile genetic elements with geNomad

翻译:周之超@UW-Madison
Nature Biotechnology, [IF 46.9]
DOI:10.1038/s41587-023-01953-y
原文链接:https://www.nature.com/articles/s41587-023-01953-y
第一&通讯作者:Antonio Pedro Camargo
通讯作者: Nikos C. Kyrpides
主要单位:
DOE Joint Genome Institute, Lawrence Berkeley National Laboratory, Berkeley, CA, USA
美国能源部联合基因组研究所,劳伦斯伯克利国家实验室,加利福尼亚州伯克利,美国
- 摘 要 -
识别和描述测序数据中的移动遗传元素对于理解它们的多样性、生态学、生物技术应用和对公共卫生的影响至关重要。在这里,我们介绍了一种名为 geNomad 的分类和注释框架,它结合了基因内容信息和深度神经网络来识别质粒和病毒的序列。geNomad 使用了一个包含超过20万个标记蛋白质文件的数据集,以提供功能基因注释和病毒基因组的分类归属。geNomad 还使用条件随机场模型,高精度地检测整合到宿主基因组中的噬菌体。在基准测试中,geNomad 在多样化的质粒和病毒分类性能方面表现出色(马修斯相关系数分别为77.8%和95.3%),显著优于其他工具。利用 geNomad 的速度和可扩展性,我们处理了超过2.7万亿碱基对的测序数据,发现了通过 IMG/VR 和 IMG/PR 数据库可获得的数百万病毒和质粒。geNomad 可在 https://portal.nersc.gov/genomad 获取。
Identifying and characterizing mobile genetic elements in sequencing data is essential for understanding their diversity, ecology, biotechnological applications and impact on public health. Here we introduce geNomad, a classification and annotation framework that combines information from gene content and a deep neural network to identify sequences of plasmids and viruses. geNomad uses a dataset of more than 200,000 marker protein profiles to provide functional gene annotation and taxonomic assignment of viral genomes. Using a conditional random field model, geNomad also detects proviruses integrated into host genomes with high precision. In benchmarks, geNomad achieved high classification performance for diverse plasmids and viruses (Matthews correlation coefficient of 77.8% and 95.3%, respectively), substantially outperforming other tools. Leveraging geNomad’s speed and scalability, we processed over 2.7 trillion base pairs of sequencing data, leading to the discovery of millions of viruses and plasmids that are available through the IMG/VR and IMG/PR databases. geNomad is available at https://portal.nersc.gov/genomad.
- 软件内容 -
geNomad 是一个从核苷酸序列中识别病毒和质粒基因组的工具。它提供了最先进的分类性能,并可用于快速从基因组、宏基因组或宏转录组中找到移动遗传元素。
速度
geNomad 的处理速度显著快于类似工具,可用于处理大型数据集。
分类归属
识别出的病毒被归属到遵循最新国际病毒分类委员会(ICTV)分类体系的分类谱系中。
功能注释
使用 geNomad 的标记数据库,对病毒和质粒编码的基因进行功能性注释。
① 软件构架和原理
混合分类框架和得分聚合 geNomad 采用一种混合方法来识别质粒和病毒,该方法结合了一种基于神经网络的分类器,它无需对参考数据库进行序列比对,以及一种基于标记的分类器,根据具有分类信息的蛋白质标记的存在对序列进行分类。为了提高分类性能,geNomad 通过将这两种模型的输出聚合为单一分类,利用了两者的优势。
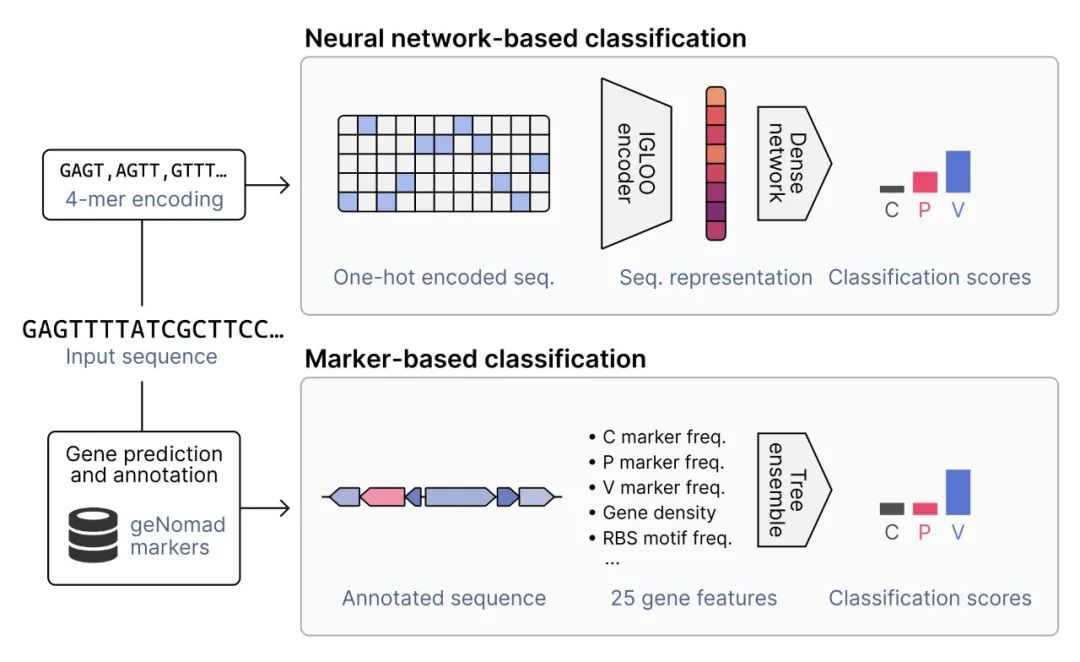
基于注意力机制的得分聚合
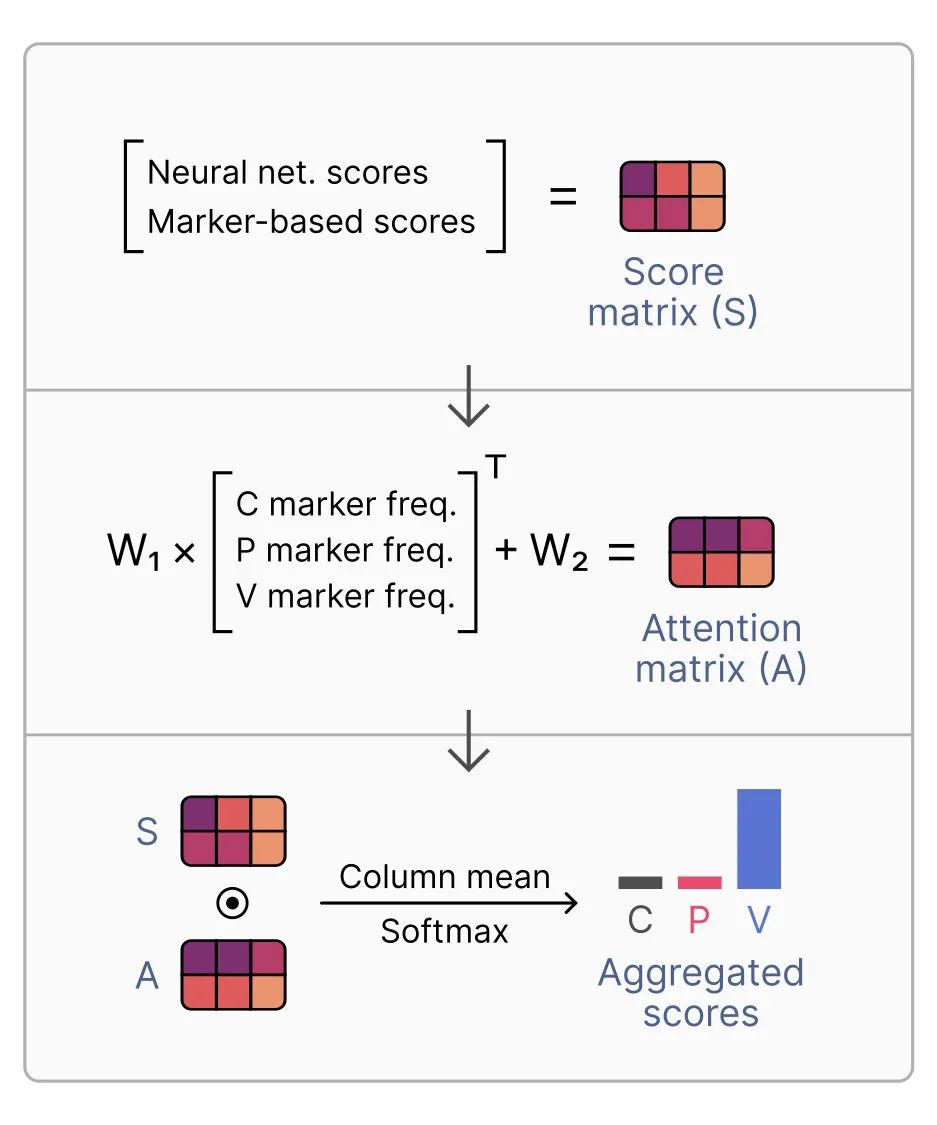
基于神经网络的分类器和基于标记的分类器使用不同且通常互补的方法来对输入序列进行分类。通过聚合它们的输出,geNomad 可以利用这两种方法的优势,从而提供更好的分类性能。这是通过一个注意力机制实现的,该机制包括一个线性模型,根据输入序列中染色体、质粒和病毒标记的频率对分支进行加权。
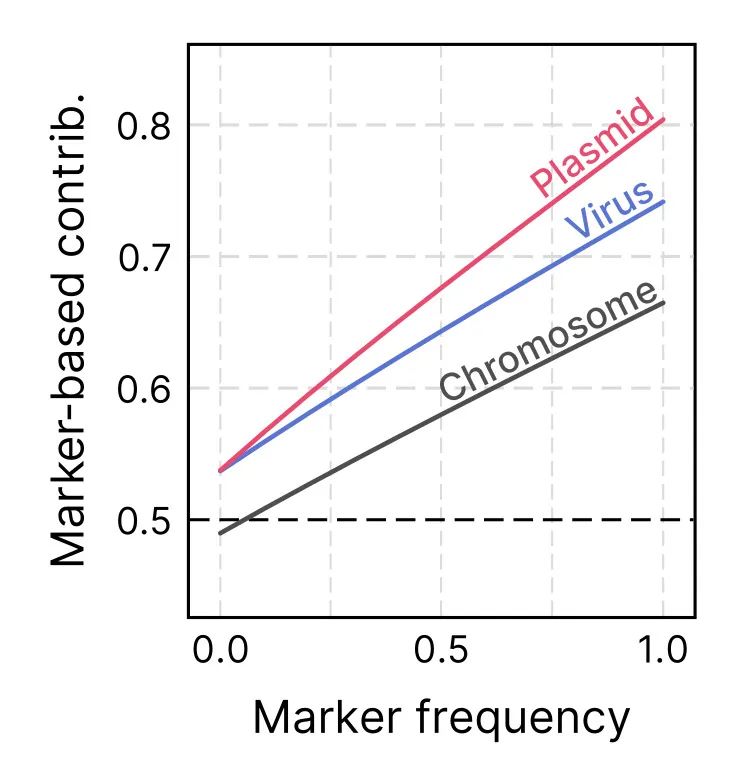
注意力机制的工作方式是,随着被分配给标记的基因比例的增加,标记分支的贡献度也随之增高。本质上,分支聚合在标记分支更具信息量时(即输入序列编码的大多数基因被分配给标记时)给予标记分支更多的权重,并在标记信息稀缺时更多地依赖序列分支。
② 基于标记的分类特征
为了使用基于标记的分类器将特定序列分类为染色体、质粒或病毒,geNomad 使用 pyrodigal-gv 进行基因预测,并使用 MMseqs2 将预测出的蛋白质分配给 geNomad 的标记。从序列的基因结构、RBS(核糖体结合位点)基序以及分配给其蛋白质的标记的身份出发,计算出一系列数值特征,并将这些特征作为分类模型的输入。
geNomad的标记数据集
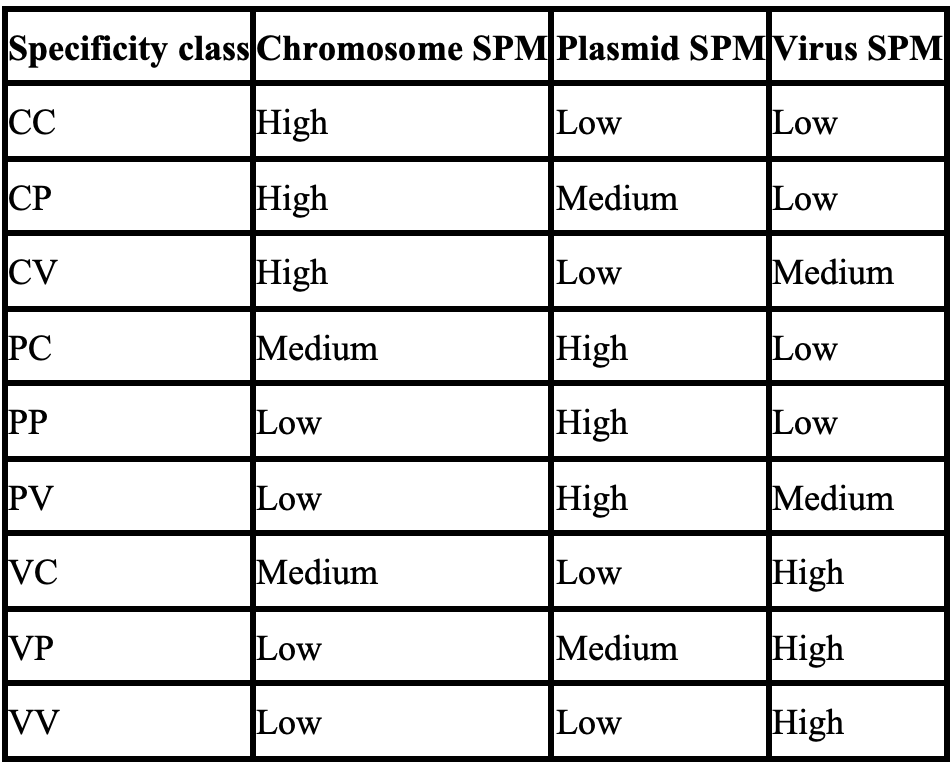
用于分类的大多数特征是根据分配给序列编码的基因的标记的特异性概况计算出来的。geNomad 的227,897个标记中的每一个都有三个相关的SPM(特异性概率测量)值,这些值从0到1变化,用于衡量该标记对三个类别(染色体、质粒和病毒)的特异性。例如:

GENOMAD.109995.PP 标记非常特异于质粒,因为其质粒SPM接近1,而其染色体和病毒SPM值接近0。另一方面,GENOMAD.109996.VV 标记显示出对病毒的高特异性,具有高病毒SPM和低染色体及质粒SPM值。所有 geNomad 标记的特异性分布在下面的三元图中表示。每个标记(圆圈)的位置由其特异性决定,颜色代表在图表特定区域的标记密度。靠近三角形顶点的标记强烈特异于其中一个类别。
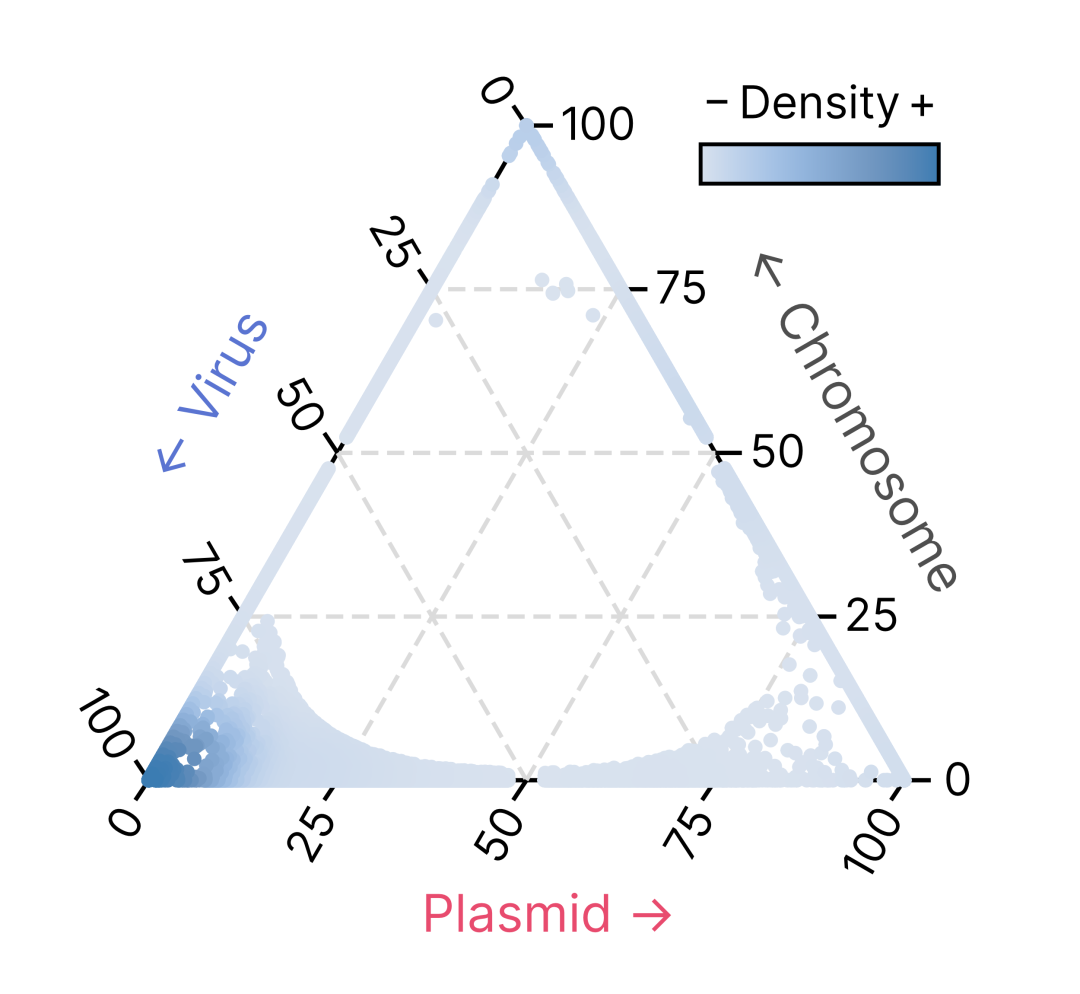
基于它们的特异性概况,标记被分配到九个类别之一:
③ 基于神经网络的分类
神经网络可以用于直接从核苷酸序列进行分类,无需明确对参考基因组或蛋白进行序列比对。这是因为某些神经网络架构能够学习对分类具有信息量的鉴别性序列基序。在 geNomad 中,基于神经网络的分类器与基于标记的分类器结合使用,以提高对分配给标记的基因较少或没有的序列的分类效果。
geNomad 的神经网络分类器

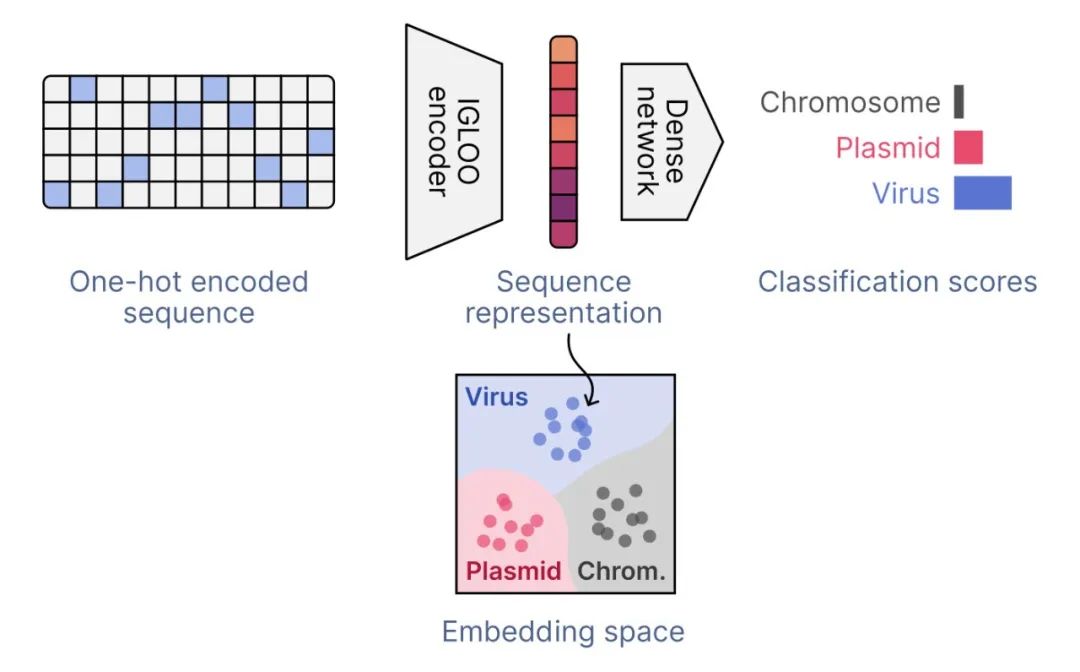
为了在不明确对参考数据库进行序列比对的情况下识别质粒和病毒的序列,geNomad 使用神经网络模型处理输入,仅通过其核苷酸组成就能对序列进行分类。为了实现这一点,输入序列首先通过将其切分为4-mer词的数组来转换为数值格式,然后进行独热编码(one-hot-encoding),创建反映特定4-mer(行)在序列不同位置(列)中存在情况的二进制256维矩阵。
这些矩阵随后被传递给一个编码器,该编码器在高维嵌入空间中生成序列的向量表示。在这个空间中,来自相同类别(染色体、质粒或病毒)的序列表示将与来自不同类别的序列表示相比更为相似。得到的表示随后被输入到一个密集神经网络中,该网络产生分类得分。
IGLOO 架构
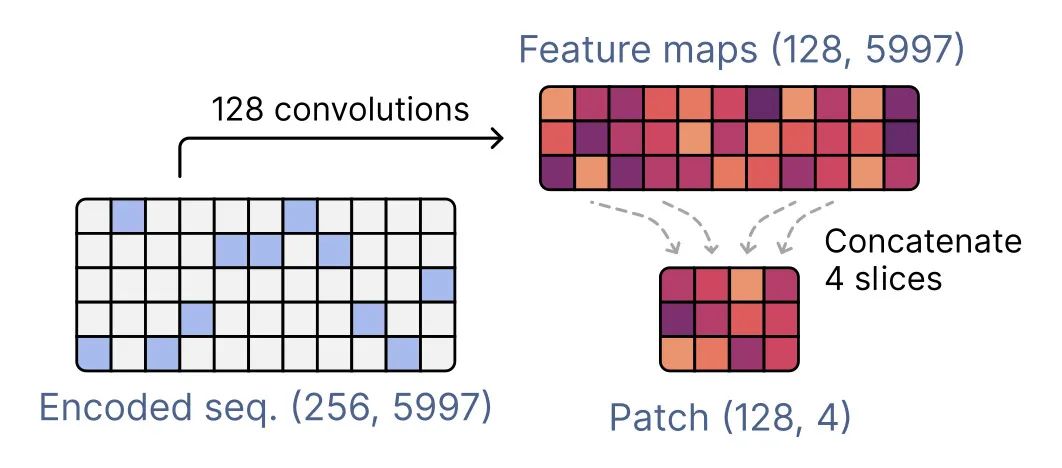
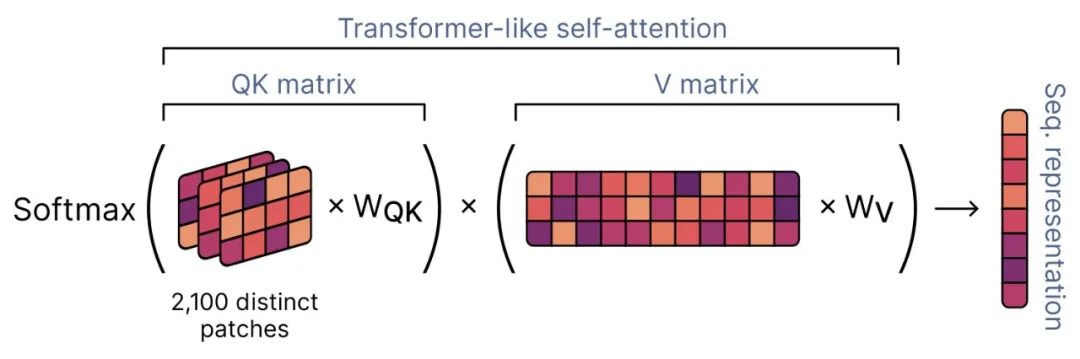
为了生成输入的向量表示,geNomad 使用基于 IGLOO 架构的编码器,该架构能够从序列数据中提取对分类有用的模式,并将它们编码到嵌入空间中。IGLOO 编码器开始处理独热编码矩阵时,通过应用卷积滤波器来生成序列特征图。为了捕捉序列非连续部分之间的关系,IGLOO 生成包含从序列内随机位置取出的片段的补丁。
这些补丁被整合到一个自注意力机制中,该机制对特征图的不同部分进行加权,并利用补丁中编码的长程依赖性来导出最终的序列表示。
④ 噬菌体整合识别
温和型噬菌体可以整合入宿主基因组,形成前噬菌体,这对宿主的代谢和生态有重大影响。geNomad 能够估算整合入宿主序列中的前噬菌体的坐标,为用户提供宝贵的信息。
前噬菌体划分算法
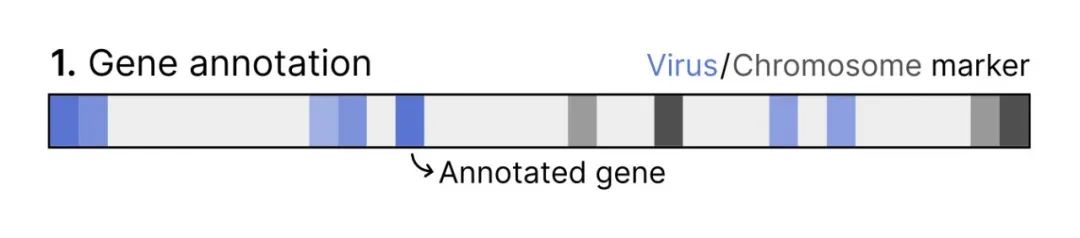
geNomad 的标记数据集提供了关于每个标记特异于病毒或宿主的具体信息。为了识别潜在的前噬菌体,geNomad 识别包含病毒特异性标记区域且被宿主特异性基因包围的序列。因此,该过程的第一步是将给定序列编码的基因分配给 geNomad 标记。
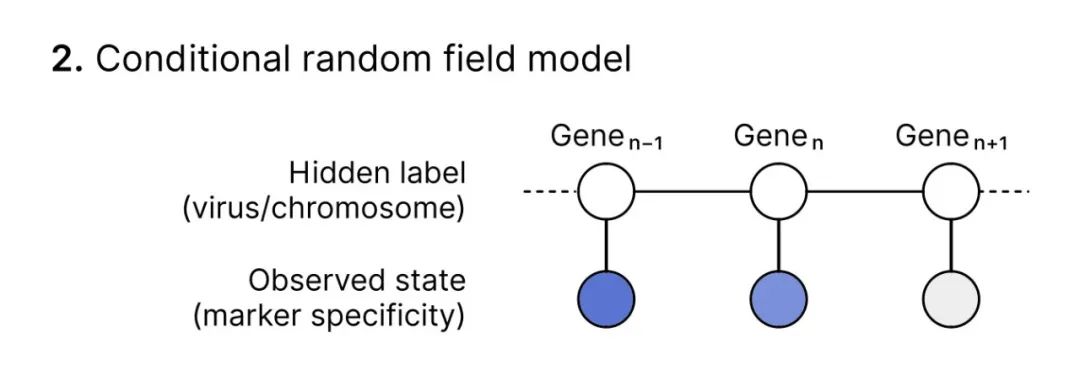
为了划分可能对应于前噬菌体的区域,geNomad 使用了条件随机场(CRF)模型。这个模型将带有 geNomad 标记的基因的染色体和病毒SPM值作为输入,并使用上下文信息来计算序列中一系列状态(染色体或前噬菌体)的条件概率。
因此,CRF 模型为每个基因提供了其属于前噬菌体的概率。
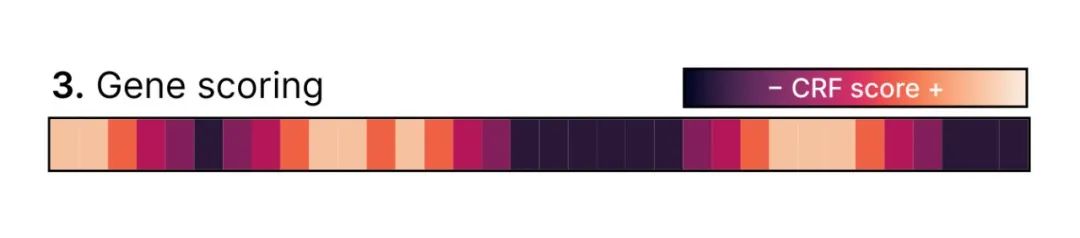
然后将基因分配到它们最可能的状态,形成前噬菌体岛屿,这些岛屿代表了富含病毒标记的区域。
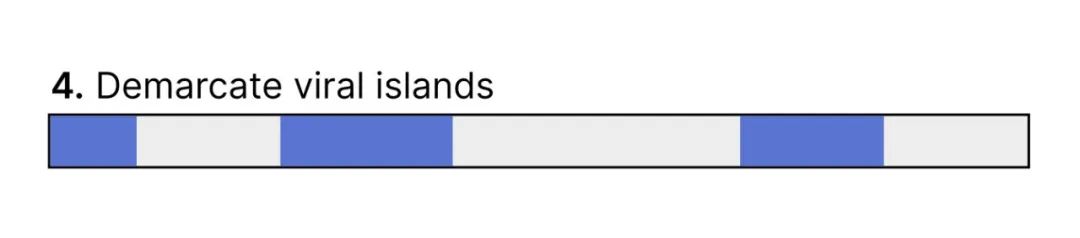
为了防止由于标记覆盖不完全导致前噬菌体被分割成多个岛屿,被短基因阵列(少于6个基因或2个染色体标记)分隔的前噬菌体岛屿会被合并。接下来,计算每个岛屿的总病毒富集度如下:
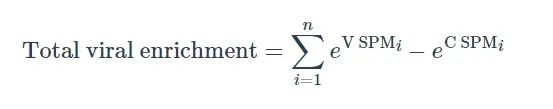
这些值代表了岛屿的总病毒富集度,考虑到了其中的所有基因。具有多个病毒特异性标记的岛屿将有更高的标记富集度,而具有少量病毒特异性基因的岛屿将有较低的标记富集度。

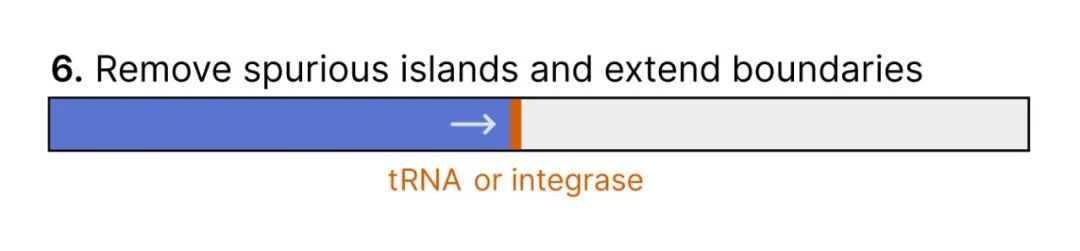
在识别出病毒岛屿后,geNomad 使用tRNA和整合酶注释来精确划定它们的边界。这是因为由于位点特异性重组动态,tRNA和整合酶通常位于整合元素旁边。因此,geNomad 将前噬菌体的边界扩展到相邻的tRNA(使用 ARAGORN 识别)和/或整合酶(使用 MMseqs2 识别,使用一组16个位点特异性酪氨酸整合酶的文件)。最后,标记富集度低的岛屿被过滤掉,因为它们通常不是真正的前噬菌体。
⑤ 病毒基因组的分类分配
geNomad 能够将病毒基因组分配到国际病毒分类委员会(ICTV)第19号病毒基因组识别标志(VMR)定义的分类谱系中。这得益于与病毒分类相关的 85,315 个标志,覆盖了 ICTV 所认可的大多数分类谱系。
分类分配算法
为了将病毒序列分配到特定的分类谱系中,geNomad 首先将序列编码的基因与 227,897 个标志的集合进行比对。产生显著匹配的基因然后被分配给一个标志,该标志可能包含分类信息(下方着色的基因)。
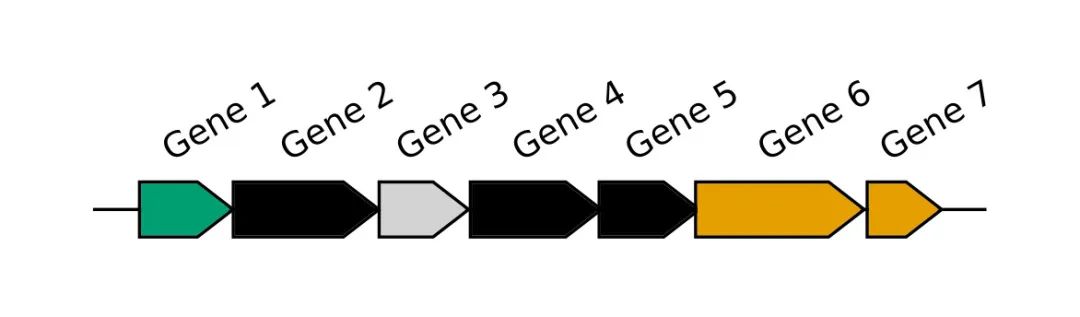
每个基因随后根据分配的标志的分类谱系进行分类。序列中的不同基因可能分配给不同的谱系。
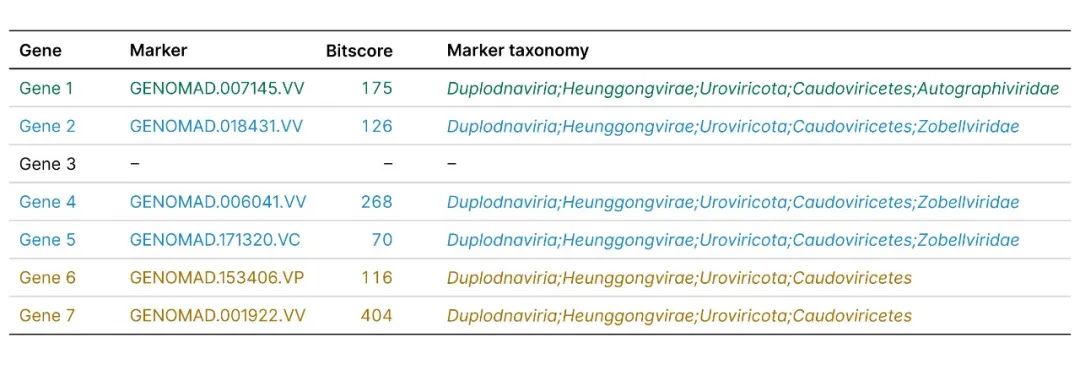
为了建立一个单一的序列级别分类体系,通过将与标志档案的比对中获得的比特分数相加,计算了包括基因级别分配中的每个分类单元以及它们的父级分类单元(直到分类体系的根部)的权重。
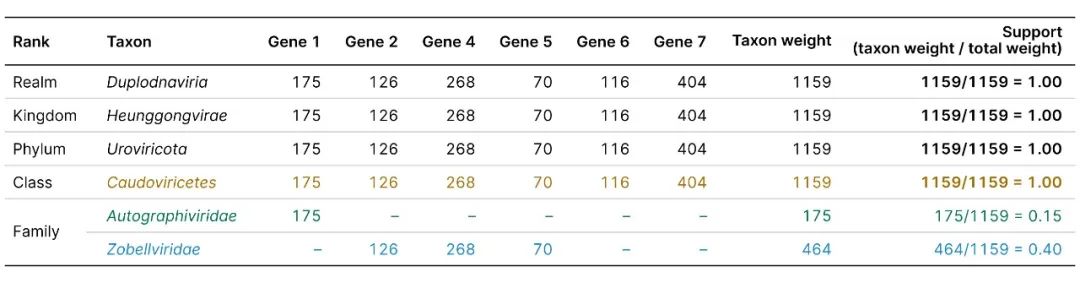
序列的分类是根据至少有总权重的50%支持的最具体的分类单元确定的(总权重是具有分类信息的所有基因的比特分数之和)。在上面的示例中,两个家族(Autographiviridae 和 Zobellviridae)都未达到50%的共识,因此该基因组被分配到 Caudoviricetes 纲。
最终分类:Duplodnaviria;Heunggongvirae;Uroviricota;Caudoviricetes。
⑥ 得分校准
geNomad的分类模型生成的分数表示它们对其预测的信心程度。然而,这些分数并不代表预测正确的真实概率。例如,如果使用相同的分类模型来识别宏基因组中的病毒(其中细胞序列数量超过病毒序列)和病毒组(富含病毒序列),预计模型在宏基因组中将产生更多的假阳性病毒,因为宏基因组中会存在更多的细胞序列(容易被误分类为病毒)。
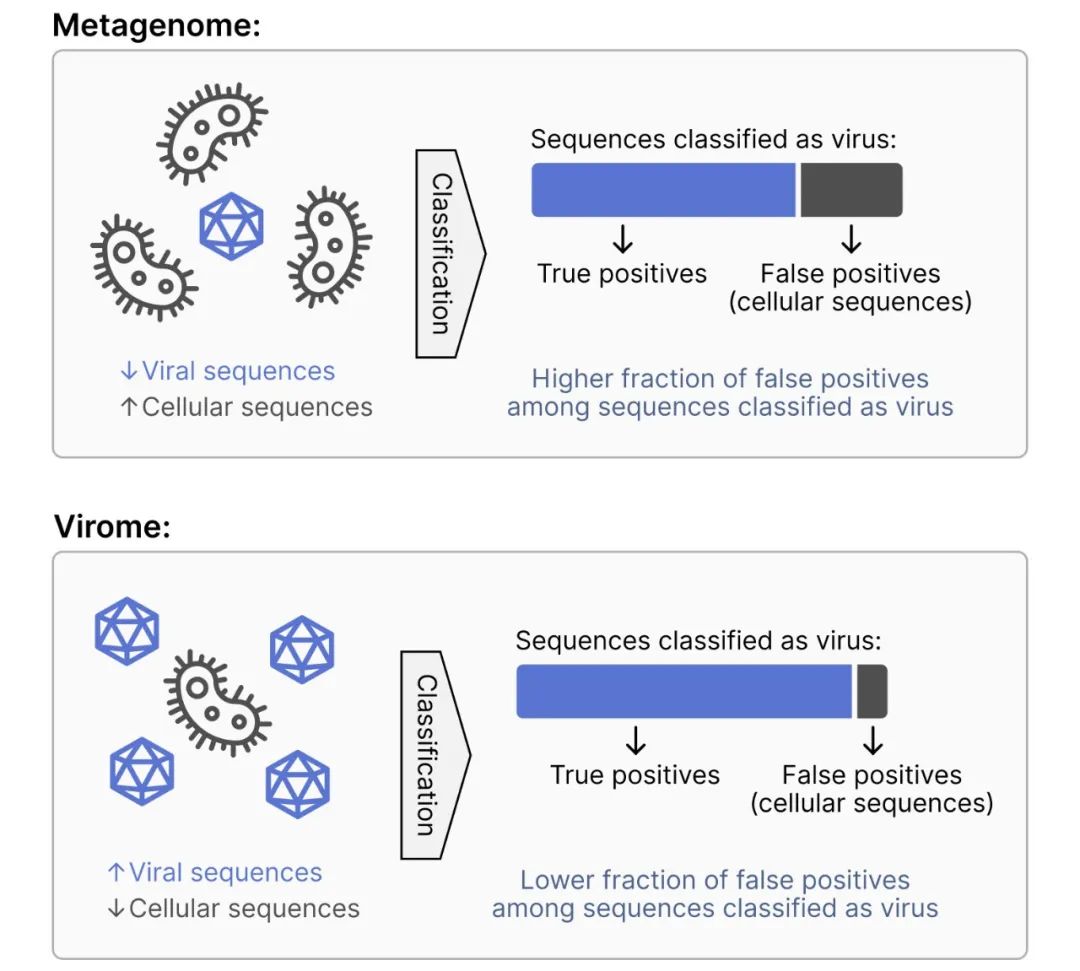
这是因为模型会为给定的序列分配相同的分数,不考虑样本的组成。为了解决这个问题,geNomad 提供了一个可选的校准机制,该机制使用样本组成数据来估计真实的概率情况。这个功能可以通过使用端到端命令的 --enable-score-calibration 参数来启用
⑦ 后分类过滤
为了识别一组序列中的质粒和病毒,geNomad 使用分类模型将序列分类为染色体、质粒或病毒。然而,仅依赖于模型的输出可能存在问题。例如,较短的序列(例如少于2,500 bp)难以分类,经常会被错误分类,因此需要额外谨慎。此外,不同的用户可能对结果有不同的期望;有些人可能希望尽可能多地预测,而其他人可能更喜欢只接受最有力支持的预测。为了应对这些挑战,geNomad 对分类结果应用了一系列过滤器。这确保了用户获得可靠的预测,同时允许他们控制过滤过程的严格程度水平。
以下过滤器可供用户使用,用于生成最终的质粒和病毒列表:
min-score: 用于标记序列为病毒或质粒的最小分数。
max-fdr: 最大接受的误差发现率。如果分数未经校准,此选项将被忽略。
min-plasmid-marker-enrichment: 质粒标志富集分数的最小允许值。
min-virus-marker-enrichment: 病毒标志富集分数的最小允许值。
min-plasmid-hallmarks: 已识别质粒中质粒标志的最小数量。
min-plasmid-hallmarks-short-seqs: 已识别质粒中长度小于2,500 bp的质粒标志的最小数量。
min-virus-hallmarks: 已识别病毒中病毒标志的最小数量。
min-virus-hallmarks-short-seqs: 已识别病毒中长度小于2,500 bp的病毒标志的最小数量。
max-uscg: 在病毒或质粒中允许的最大通用单拷贝基因(USCGs)数量。
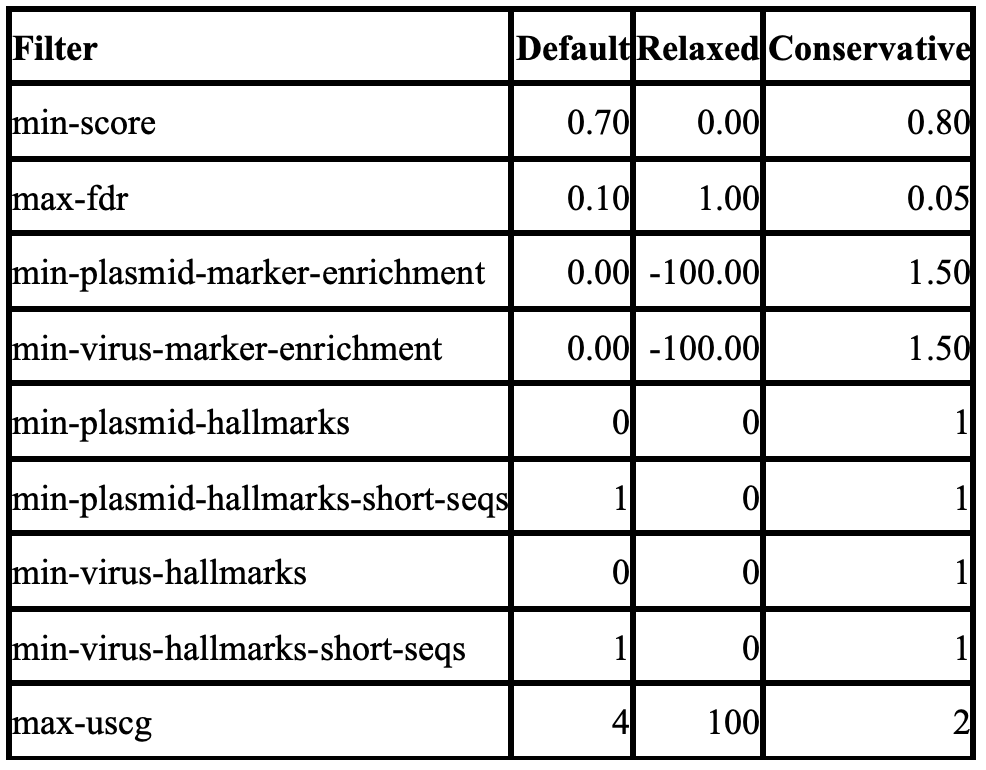
默认参数和预设值
鉴于可用的大量过滤器,geNomad 还允许用户使用预设值,可以禁用所有过滤器(--relaxed)或使过滤过程更为严格(--conservative)。
在使用默认参数或其中一个预设值执行 geNomad 时,用于过滤预测的值如下:
软件的使用和结果的解读
请参考:https://portal.nersc.gov/genomad/quickstart.html
参考文献
Camargo, A.P., Roux, S., Schulz, F. et al. Identification of mobile genetic elements with geNomad. Nat Biotechnol (2023). https://doi.org/10.1038/s41587-023-01953-y
- 拓展阅读 -
iMeta |
威斯康星大学周之超开发模块化工具研究病毒组
https://mp.weixin.qq.com/s/OVD6G9siN8eXrH1lijU-nw
GitHub: https://github.com/AnantharamanLab/ViWrap
paper: https://onlinelibrary.wiley.com/doi/full/10.1002/imt2.118
视频解读:
Bilibili:https://www.bilibili.com/video/BV16k4y1p7T3/
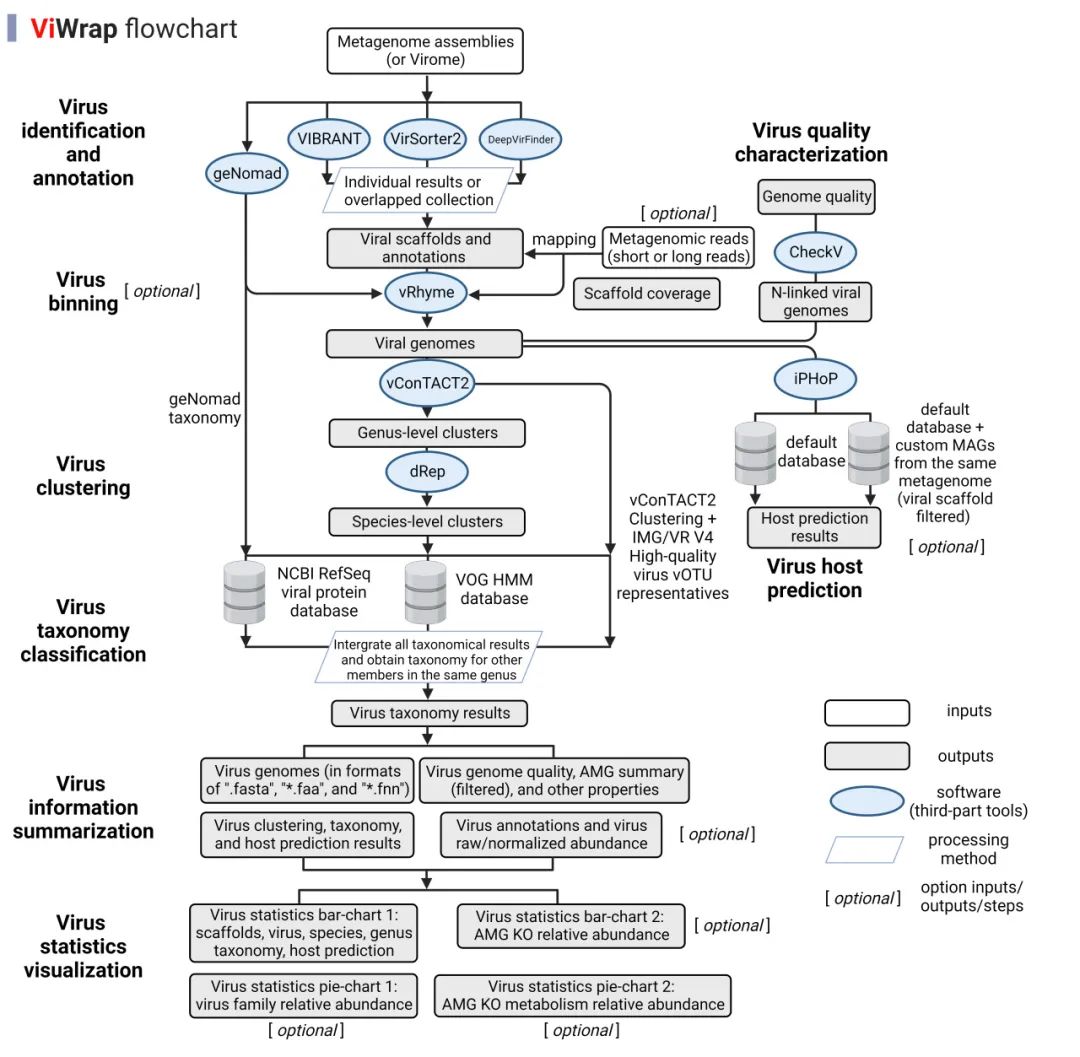
ViWrap v1.3.0 版本更新说明
在 ViWrap v1.3.0 版本的更新中,我们整合了最新版本的 geNomad v1.7.4。凭借 geNomad 在病毒识别领域的卓越性能,ViWrap 将 geNomad 设为默认病毒识别工具。为了配合新病毒识别工具的加入,本次更新包括以下几个重要内容:
1. 病毒蛋白注释的全面性提升:我们结合 VIBRANT 和 geNomad 的庞大数据库,对病毒蛋白进行了全面而详尽的注释。
2. 分类信息的精确性增强:应用 geNomad 的 “--conservative-taxonomy” 选项,我们提供了相比前版本更为全面和精确的分类信息。
3. AMG 过滤机制:新版本增加了 AMG 过滤步骤。通过这一步骤,我们删除了位于 contig 末端、具有显著病毒特征倾向、被细胞来源基因包围、以及属于 T/B 类别 COG 的 AMG,从而提供了经过筛选的 AMG。
4. iPHoP 分析的优化:对用户输入的 custom MAG 进行了进一步的过滤处理,有效清除 MAG 中可能存在的病毒 scaffold以免得到假阳性结果。
以上更新旨在提升 ViWrap 的性能和使用效率,确保用户能够更准确、高效地进行病毒识别和分析。
- 通讯作者简介 -

能源部联合基因组研究所
Nikos C. Kyrpides
原核生物超级项目负责人
简介:
Kyrpides博士于2004年加入能源部联合基因组研究所(DOE Joint Genome Institute),领导基因组生物学项目和微生物基因组和元基因组的比较分析平台的开发(IMG)。他在2010年成为元基因组学项目负责人,并从2011年起负责微生物基因组和元基因组的合并项目。在加入能源部联合基因组研究所之前,Kyrpides博士在伊利诺伊州芝加哥的Integrated Genomics Inc.领导基因组分析和生物信息学核心部门的发展。他在伊利诺伊大学厄巴纳-香槟分校和阿贡国家实验室跟随Carl Woese(古菌发现者)进行了博士后研究。Kyrpides博士的研究重点是微生物组研究,重点是微生物组数据科学。他的小组正在开发新的方法,以实现大规模的比较分析,以及大数据的挖掘和可视化。
主页:
https://jgi.doe.gov/our-science/scientists-jgi/nikos-kyrpides/

能源部联合基因组研究所
Antonio Camargo
项目科学家
简介:
安东尼奥的研究专注于开发新方法,用于从宏基因组数据中识别病毒序列。他是IMG/VR4 (病毒)和IMG/PR (质粒)数据库以及软件geNomad和RNA virus annotation pipeline (RVAP) v1.0的主要作者。
主页:
https://jgi.doe.gov/our-science/scientists-jgi/microbiome-data-science/
宏基因组推荐培训/会议
猜你喜欢
iMeta高引文章 fastp 复杂热图 ggtree 绘图imageGP 网络iNAP
iMeta网页工具 代谢组MetOrigin 美吉云乳酸化预测DeepKla
iMeta综述 肠菌菌群 植物菌群 口腔菌群 蛋白质结构预测
10000+:菌群分析 宝宝与猫狗 梅毒狂想曲 提DNA发Nature
一文读懂:宏基因组 寄生虫益处 进化树 必备技能:提问 搜索 Endnote
16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流快速解决科研困难,我们建立了“宏基因组”讨论群,己有国内外6000+ 科研人员加入。请添加主编微信meta-genomics带你入群,务必备注“姓名-单位-研究方向-职称/年级”。高级职称请注明身份,另有海内外微生物PI群供大佬合作交流。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍未解决群内讨论,问题不私聊,帮助同行。
点击阅读原文,跳转最新文章目录阅读

















 1280
1280

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









