






DrugGPS是 一种基于亚口袋和分子motif的分子生成方法,是中科大计算机学院刘淇教授 ICML 2023 的文章 《Learning Subpocket Prototypes for Generalizable Structure-based Drug Design》。

一、模型介绍
DrugGPS( a structure-based Drug design method that is Generalizable with Protein Subpocket prototypes)是针对口袋条件下的,基于分子motif的3D分子生成网络。其模型结构如下图:
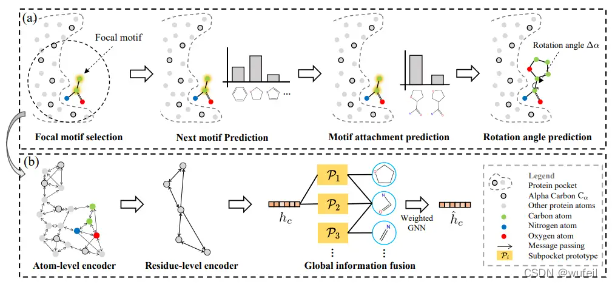
DrugGPS是基于他们之前开发的FLAG方法,与FLAG类似,分子生成过程是一个 motif-by-motif的过程,仍然是包含了局部motif选择,下一个motif预测,motif的连接位置预测,motif链接之后的扭转角预测。
不一样的是,在嵌入motif的时候,在encoder中,使用了原子层面和残基层面的3D graph transformer(如上图b),以捕捉分层的结构信息。然后构建了一个全局相互作用图,去模拟子口袋和配体之间的相互作用。该全局的相互作用图有两种节点:子口袋类型节点和motif库中分子motif节点。如果在数据集中,同一类的子口袋与某个motif之间存在相互作用,那么在他们之间添加一条边,如果出现的频率越高,则边的权重越大。关于相互作用的判断,作者使用的BINANA工具,仅计算口袋类型和motif之间的氢键相互作用。关于全局相互作用图的边的权重更新方法,如下图,详看作者文章3.4 Global Interaction Graph Construction部分。

因此,可以认为DrugGPS是FLAG的改良模型,在算法上改良了口袋和分子motif之间的相互联系,在数据上,新增了H键相互作用。
DrugGPS可以识别motif和子口袋类型之间的相互作用,故能生成结合力更强的分子。全局的相互作用图是之前FLAG方法没有的。
二、文章结果介绍
作者对比了DrugGPS和FLAG, Pocket2Mol等模型的对比结果,如下图:
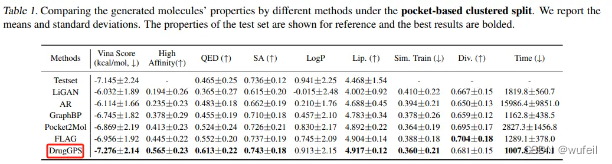
可以看到DrugGPS生成分子的vina score,要明显优于FLAG等模型。此外,DrugGPS生成的分子,在成药性,可合成性上也优于之前的模型,效果比较明显。
作者还研究了子口袋类型数量与motif数量对生成分子vina score之间的关系,如下图。
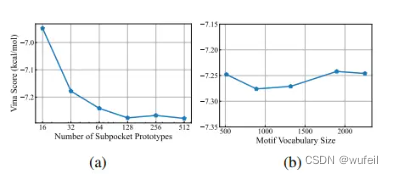
还有,就是一些消融实验。
可能是因为预印本的原因,代码可能仍在修改,实验的结果并不多。
三、模型测评
作者在文章中提供了GitHub 链接:GitHub - zaixizhang/DrugGPS_ICML23: Implementation of ICML23 paper "Learning Subpocket Prototypes for Generalizable Structure-based Drug Design"
首先,复制项目到本地
git clone https://github.com/zaixizhang/DrugGPS_ICML23.git安装conda环境
conda env create -f druggps_env.yaml
conda activate druggps_env
pip install kmeans-pytorch由于当前作者并未提供已经训练好的checkpoint和处理好的数据集,因此,需要数据预处理开始。
1. 下载数据集
作者使用的是crossdocked_pocket10.tar.gz数据集。下载链接为:
https://drive.google.com/drive/folders/1CzwxmTpjbrt83z_wBzcQncq84OVDPurM
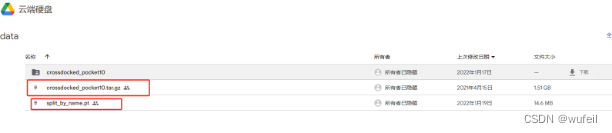
里面的两个文件都要下载。下载完成后,放置在./data文件夹内,然后解压crossdocked_pocket10.tar.gz文件。
tar -xzvf crossdocked_pocket10.tar.gz然后切换到 ./utils目录,执行如下命令,为模型提取H键相互作用的原子:
cd utils
unzip binana.zip
python preprocess_data.py注:unzip binana.zip 解压binana模块,否则会报错:ModuleNotFoundError: No module named 'binana'。
此外,在执行上述命令过程中,发现应该会生成一个atoms_hbond.npy文件,用于记录每个口袋-分子对中H键原子。
但是,由于作者提供的数据集中,没有pdbqt文件,而作者使用的提取H键相互作用的程序是binana是需要pdbqt文件作为输入的,导致无法提取H键相互作用原子。
因此,直接运行上述命令,生成的atoms_hbond.npy实际上是空值,不影响后面的训练,相当于没有相互作用。
我单独为crossdocked_pocket10中的分子和蛋白生成pdbqt文件,然后再进行预处理。
成功生成了含有H键的数据集。为了与代码中的路径配合,需要将atoms_hbond.npy需要移动到./目录下。
2. 训练模型
由于作者没有提供训练好的模型checkpoint,我们只能自己训练。
按照作者在github上的介绍,在./目录下执行如下命令:
python train.py执行时,可能会遇上如下问题,包括:
(1)FileNotFoundError: [Errno 2] No such file or directory: './atoms_hbond.npy',请调整./atoms_hbond.npy文件放置在./目录下。
(2)ModuleNotFoundError: No module named 'chemutils'。这个可能是执行路径的原因,改一下代码就好,在train.py文件中,添加
import sys
sys.path.append("..")
sys.path.append("./utils") # wufeil(3)FileNotFoundError: [Errno 2] No such file or directory: './data/crossdocked_pocket10_name2id.pt',删除./data文件夹中的crossdocked_pocket10_processed.lmdb;从新执行上述训练代码就可以生成了。
这是因为加载数据集时,如果存在crossdocked_pocket10_processed.lmdb文件,就认为已经存在处理过的数据集,就不再重新处理,所以直接加载。直接加载需要每个蛋白的index文件,即crossdocked_pocket10_name2id.pt,但是作者没有提供。
因此,删除crossdocked_pocket10_processed.lmdb文件重新运行即可。
但是后续运行中,加载数据集报错,所有的口袋-分子都无法加载到模型中。其实,这里这么处理是不对的,因为作者使用了H-bond相互作用,分子的序号是不一样的。
!!!关于这一部分,我查看了原始代码,并进行了修改,修改内容复杂,就不描述了。具体原因是在处理分子的时候,parse_sdf_file函数中 moltree = MolTree(mol)少了一个参数。
总的来说,改代码改的很累很累,小错误太多,很有可能是我们的版本问题。
因此代码中的小修改太多了,这里就不一一列举了。
在更改代码以后,重新运行
python train.py会自动按照配置文件,开始处理数据集,生成crossdocked_pocket10_processed.lmdb和crossdocked_pocket10_name2id.pt。运行输出如下:
在长时间的数据处理之后(大约是6个小时),开始执行训练,但是报错
File "/home/DrugGPS/models/protein_features.py", line 47, in _rbf
RBF = torch.exp(-((D_expand - D_mu) / D_sigma)**2)
RuntimeError: Expected all tensors to be on the same device, but found at least two devices, cuda:0 and cpu!此时,仍然需要代码。修改过程较为复杂,这里就不描述了。在修正代码后,开始训练,输出每个批次的训练损失:
每迭代10000次保存一次模型checkpoint。
如果训练过程中遇到,如下错误:
File "train.py", line 188, in proto_init
_, centers = kmeans(X=torch.cat(emb_list, dim=0), num_clusters=config.train.num_prototypes, iter_limit=100)
TypeError: kmeans() got an unexpected keyword argument 'iter_limit'是因为kmeans_pytorch版本的原因,需要重新安装一下。安装指定版本,命令如下:
sudo pip3 install -e git+https://github.com/subhadarship/kmeans_pytorch.git#egg=kmeans_pytorch训练开始后,会在log文件夹内生成一个对应时间的文件夹,例如:train_model_2024_01_24__16_45_04,其中的文件目录如下:
.
├── checkpoints
├── events.out.tfevents.1706089504.a01.1834344.0
├── log.txt
├── models
└── train_model.yml
2 directories, 3 files其中,models文件夹为对应的模型代码,log.txt为训练过程输出,train_model.yml为训练过程中的超参数配置,checkpoints为训练过程中的pt格式的checkpoint,即模型权重。我最终一共训练了600000次迭代,3090显卡花费了5天时间,获得了一个600000.pt的checkpoint。
3. 生成分子
首先将我们训练获得的600000.pt从/logs/train_model_2024_01_24__04_45_04/checkpoints目录拷贝出来,放置在./目录下。
然后,修改分子采样配置文件,修改为(其实在训练模型时,也作了修改。。。):
dataset:
name: pl
path: ./data/crossdocked_pocket10
split: ./data/seq_split.pt
model:
hidden_channels: 256
subpocket_motif: True
num_prototypes: 64
checkpoint: ./600000.pt
encoder:
name: hierGT
hidden_channels: 256
edge_channels: 64
key_channels: 128
num_heads: 4
num_interactions: 6
cutoff: 10.0
knn: 32
num_filters: 128
sample:
seed: 2023
num_samples: 100
beam_size: 10
logp_thres: -.inf
num_retry: 5
max_steps: 12
batch_size: 10
num_workers: 4按照作者早github上的描述,我们再./目录下执行如下命令:
python sample.py默认是使用CPU。运行输出为:
在生成结束以后,输出所有生成分子的SA,QED, LogP, Lipinski的平均值。,如下:
QED 0.493651 | LogP 0.448651 | SA 0.589300 | Lipinski 4.890000
这一结果显示,生成的分子类药性还可以的。
根据运行的时间,会在outputs文件夹内生成一个以时间命名的文件夹,例如:sample-0_2024_01_29__21_34_31,文件夹内包含了生成100个分子的sdf文件,和分子的smiles文件SMILES.txt,还包括口袋信息文件pocket_info.txt,以及相应的生成分子的配置文件sample.yml,所有的文件目录如下:
.
├── 0.sdf
├── 10.sdf
├── 11.sdf
├── 12.sdf
...
├── 98.sdf
├── 99.sdf
├── 9.sdf
├── log.txt
├── pocket_info.txt
├── sample.yml
└── SMILES.txt
0 directories, 104 files我们使用如下命令,将所有的生成分子的sdf文件合并成all.sdf, 在sample-0_2024_01_29__21_34_31目录下执行。
for i in `ls *.sdf | sort -t'_' -k 4,4 -n `; \
do cat $i >> all.sdf; done按照作者在代码中的设定,对测试集的第一个样本(即,crossdocked_pocket10数据集中的BSD_ASPTE_1_130_0文件夹下的2z3h_A_rec_1wn6_bst_lig_tt_docked_3_pocket10.pdb,即2z3h.pdb)进行分子生成,生成100个分子。2z3h口袋结构及其原来的配体结构如下图:
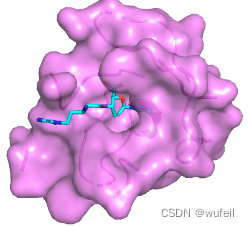
配体结构:
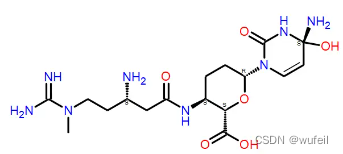
DrugGPS生成分子的2D结构如下(部分):

生成的3D分子与口袋的位置如下视频:
DrugGPS_Case_2z3h
其实这个体系,在之前的FLAG的文章中也有介绍过,DrugGPS的结果,分子更小一些,没有FLAG的大。但是与FLAG生成的分子类似的,仍有大量的并环结构存在。可惜,在测试FLAG模型时,生成的分子并没有进行对接,因此无法在这个体系上指导,是FLAG生成的分子还是DrugGPS生成的分子bind ding更好。注意,在生成的分子速度上,100个分子,DrugGPS确实非常快,大约只花了5分钟,要比FLAG快很多。
对于原来的晶体参考分子而言,DrugGPS生成的分子偏小,可能是DrugGPS仅仅针对半封闭口袋部分进行分子生成,但是对于暴露在溶剂区的位置,并未生成片段。
从生成分子与口袋的3D位置来看,生成的分子大部分都能不与口袋发生“碰撞”但是,发生碰撞的情况也非常明显。
另外,作者使用rdkit保存3D分子为sdf格式,有时会因为原子的连接顺序问题,导致出现错乱情况,如下图:
比较重要的一点是,DrugGPS生成分子含有大量的极性基团位于支链或者侧链位置,例如-OH等。这一点是非常奇怪的,这些分子显然是不类药的,但是QED score不能识别的。原因可能是:
(1)一个分子的生长迭代次数太多了,当找不到口袋内的生长点时,模型会不断尝试一些支链和侧链位置,那么就会生成一些侧链基团,这一点可以通过限制生长步数,以及生长点限制来实现。
(2)另外一种原因是,数据集中其实包含了大量的非类药的分子,特别是一些内源性分子,含有大量的OH, PO4结构,这都导致模型倾向生成极性的侧链基团。
(3)分子片段的切割方法,也是一种解决思路,特别是减少小片段。
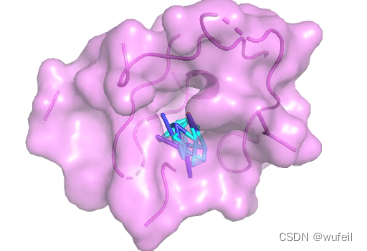
为了查看参考分子和对接分子的打分,进行了对接。
参考分子的对接打分为-8.735,对接pose与晶体中的类似,如下图:
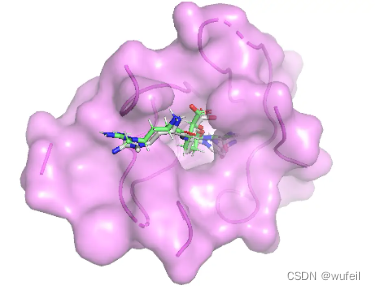
生成分子中,前三好的打分分别为:-7.78, -6.22, -6.14,对应的对接pose分别如下图:
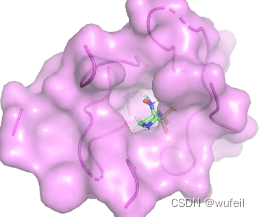
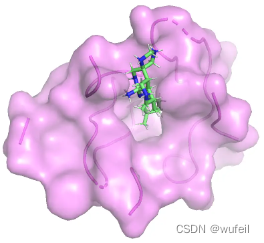
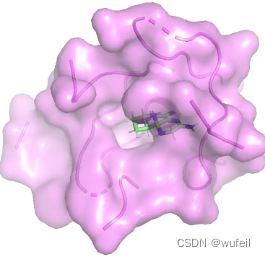
考虑到生成分子仅仅占据口袋区域,且生成分子较小,为此,计算了LE。参考分子的LE 为-0.281,上述三个分子的LE为-0.458, -0.212, -0.249。从LE的角度上看,DrugGPS确实生成了一些结合力更强的分子。
当然,模型真实的情况,需要作者提供训练好的checkpoint才能知道。
我们自己训练的模型,不知道是否迭代次数是否充足。我也没有进行超参数优化,包括epoch的选择都是直接选最后一个epoch。
就连数据集中,生成pdbqt文件然后使用BINANA计算H键,不知道是否有问题。
毕竟代码里面小错误很多,改的有点乱了。这也是没有提供这个模型修改方法的原因。
四、总结
DrugGPS是基于FLAG 改进而来,提出了一种基于亚口袋原型和motif的分子生成方法,使用原子层面和残基层面的3D graph transformer,捕捉分层的结构信息,以识别motif和子口袋类型之间的相互作用,故能生成结合力更强的分子。
经过修改作者提供的代码,成功训练迭代了600000次,然后,按照作者提供的采样代码,针对测试集中的第一个体系2z3h,进行分子生成测试。2z3h体系上一共生成了100个3D分子。生成3D分子很好落在口袋范围内,但是部分生成分子存在于口袋发生碰撞的情况,同时生成的分子较小,未占据到溶剂区。此外,生成分子中,存在大量的极性侧链基团,导致类药性差。
重对接的都docking和LE结果表明,DrugGPS生成分子与口袋之间具有较强的结合力。
DrugGPS是一次比较好的概念研究,距离实际应用还有太多需要工业界的开发。在应用模式上,需要进行改进,改为制定生长位点的指定迭代次数生长。


















 1069
1069

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











