







该推文首发于公众号:单细胞天地
上一讲在完成了从T/NK至CD4+T细胞流程实践后,在这一讲内容中我们将对CD4+T细胞进行亚群的细分。中级篇1最后的工程文件以及本次课程中会用到的一些文件:链接: https://pan.baidu.com/s/18_jO0y6_QyEtyzZJkmIp_Q 提取码: 27jw 。此外,可以向“生信技能树”公众号发送关键词‘单细胞’,直接获取Seurat V5版本的完整代码。
需要提醒的是,本次细胞亚群注释较为复杂,笔者更多会从技术角度展示分析流程和结果,而非从生物学背景角度进行解读! 具体的亚群细分命名笔者主要会从三大块进行考量:1. 先验知识证据;2. 转录因子结果;3. 差异基因结果;
首先需要对常规的CD4+T细胞亚群具有一定了解,这些细胞亚群包括了:幼稚性T细胞,辅助性T细胞(THs),记忆性T细胞和调节性T细胞(Treg)等细胞。
-
Naive CD4+T细胞:是一类尚未遇到抗原的 T 辅助细胞(T helper cells, Th)。它们在胸腺中发育成熟后进入外周免疫系统,处于静息状态,尚未被抗原刺激或激活。
-
辅助性T细胞:主要通过信号传导支持其他免疫细胞的功能。根据它们分泌的细胞因子不同,可以分为多个亚型(Th1,Th2,Th9,Th17,Th22,Tfh),每种亚型在免疫应答中发挥不同的作用。
-
记忆性T细胞:是一类能够长期存活并在二次免疫应答中迅速反应的 T 细胞。这里仅考虑CD4+记忆性T细胞。
-
调节性T细胞:对维持自身免疫耐受和免疫稳态方面至关重要,同时在某些情况下也可能促进疾病进展。其中,Foxp3 是 Treg 细胞的重要标志物。
分析步骤:
1.导入
rm(list=ls())
options(stringsAsFactors = F)
source('scRNA_scripts/lib.R')
source('scRNA_scripts/mycolors.R')
library(stringr)
library(Seurat)
library(ggplot2)
library(clustree)
library(cowplot)
library(data.table)
library(dplyr)
library(qs)
library(BiocParallel)
register(MulticoreParam(workers = 8, progressbar = TRUE))
dir.create("./9-CD4+T/")
setwd("./9-CD4+T")
sce <- qread("sub_data_cd4+T.qs")
DimPlot(sce)
table(sce@meta.data$RNA_snn_res.2)
# 0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18
# 421 410 408 390 379 373 349 333 328 328 313 293 285 280 248 204 196 189 185
2.基于先验知识的经典细胞亚群分类及注释
先按照生物学知识尝试分群,这就需要研究者有较强的生物学背景,通过人工阅读差异基因列表来判断,可以使用FindAllmarkers函数,也可以使用cosg做差异分析。(顺便提一下张泽明院士团队对差异基因的其中一个评价标准:每个簇的标志性基因是通过 Seurat软件包中的 FindAllMarkers 函数获得的,符合以下标准的基因被认为是特征基因:1. 经过 Bonferroni 校正后,所有特征基因的调整 P 值 < 0.01;2. 平均表达量的对数倍数变化(log fold-change)> 1;3. pct.1 > 0.5(即该基因在该簇中表达的细胞比例大于 50%);4. pct.2 < 0.5(即该基因在其他簇中表达的细胞比例小于 50%),这个标准可以学习借鉴。)
此外,流式分选时采用的“层层递进”策略也是值得我们学习的,但由于流式检测的是细胞表面标志物,这些标志物不适用于单细胞数据分析,因此需要更换为适合单细胞数据的标志物。
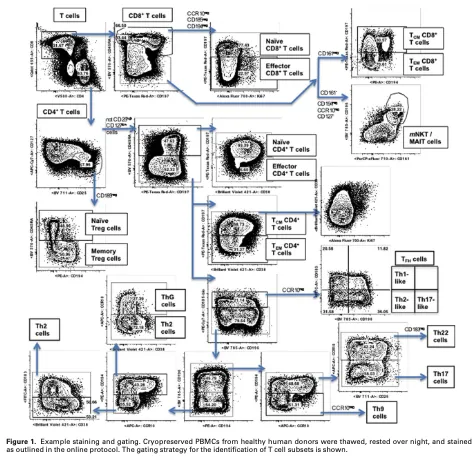
2.1 区分Treg/not Treg
为什么选择Treg,因为笔者认为这个细胞亚群与其他的细胞亚群相比,存在较大的相对异质性。同时也符合流式分选的流程。
CD4_markers_step1 =list(
T_check=c("CD3D","CD3E","CD4",'CD8A',"PTPRC"),
Treg=c("TNFRSF4","BATF","TNFRSF18","FOXP3","IL2RA","IKZF2","IL7R")
)
genes_to_check = lapply(CD4_markers_step1, function(x) str_to_upper(x))
DotPlot(sce, features = genes_to_check ) +
# coord_flip() +
theme(axis.text.x=element_text(angle=45,hjust = 1))
w=length( unique(unlist(genes_to_check)) )/5+12;w
ggsave(filename = "CD4_T_markers_step1.pdf", width = w)
# featurePlot可视化
marker <- c("PTPRC")
p <- FeaturePlot(sce,features = marker,cols = c("lightgrey" ,"red"),
combine = TRUE,raster=FALSE)
p|DimPlot(sce,label = T)
ggsave("featurePlot_step1.pdf",width = 12,height = 7)
# VlnPlot可视化
library(paletteer)
VlnPlot(sce,features = marker,cols = c(paletteer_d("awtools::bpalette"),
paletteer_d("awtools::a_palette"),
paletteer_d("awtools::mpalette"),
paletteer_d("awtools::spalette")),
combine = TRUE,raster=FALSE)
ggsave("VlnPlot_step1.pdf",width = 9,height = 7)
根据第一步结果,第3,6,10,13,16,18簇可被认为是Treg细胞。
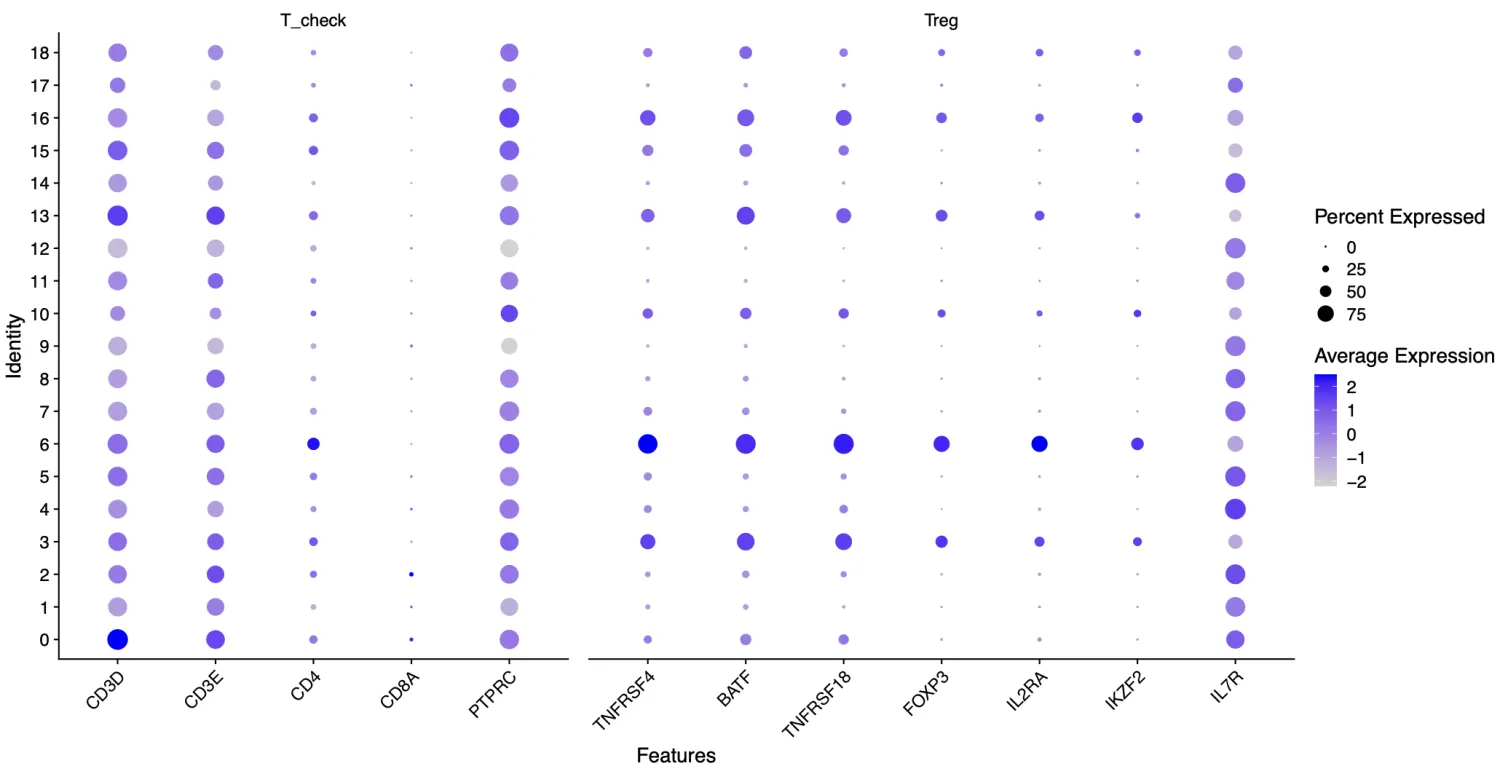
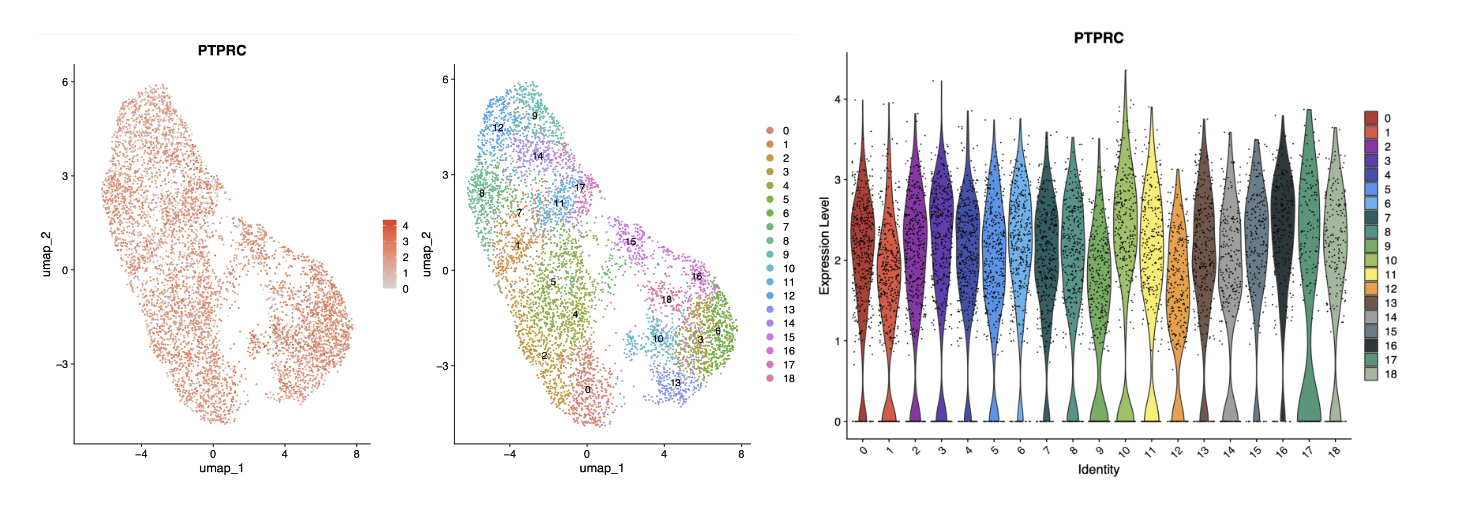
PTPRC又叫做CD45,从单细胞测序水平来看其几乎在所有细胞中都是表达的(RNA水平),而在进行流式的时候是可以用这个基因表达的阳/阴性去区分细胞亚群(蛋白质水平),因此不可完全采用流式分选中使用的标志物在单细胞数据中进行分群。
2.2 区分其他T细胞亚群
理论上也应该层层递进下去,但实操中发现还是有点难度,不同细胞亚群的标志物界限比较模糊
# 进一步区分其他亚群
CD4_markers_step2 =list(
T_check=c("CD3D","CD3E","CD4",'CD8A',"PTPRC"),
Naive=c("CCR7","SELL","CD5","LEF1","TCF7"), # CD197,CD62L
Memory=c("CXCR4","TNFRSF4","RORA","IL17RA"),
Tfh=c("CXCR5","BCL6","ICA1","TOX","TOX2","IL6ST"),#滤泡辅助性T细胞;CD185=CXCR5
Ths=c("CCR10","CCR4","CCR6","CXCR3")#"CCR10","CD194","CD196","CD183"
)
genes_to_check = lapply(CD4_markers_step2, function(x) str_to_upper(x))
DotPlot(sce, features = genes_to_check ) +
# coord_flip() +
theme(axis.text.x=element_text(angle=45,hjust = 1))
w=length( unique(unlist(genes_to_check)) )/5+12;w
ggsave(filename = "CD4_T_markers_step2.pdf", width = w)

2.3 张泽民院士团队,PMID: 30479382
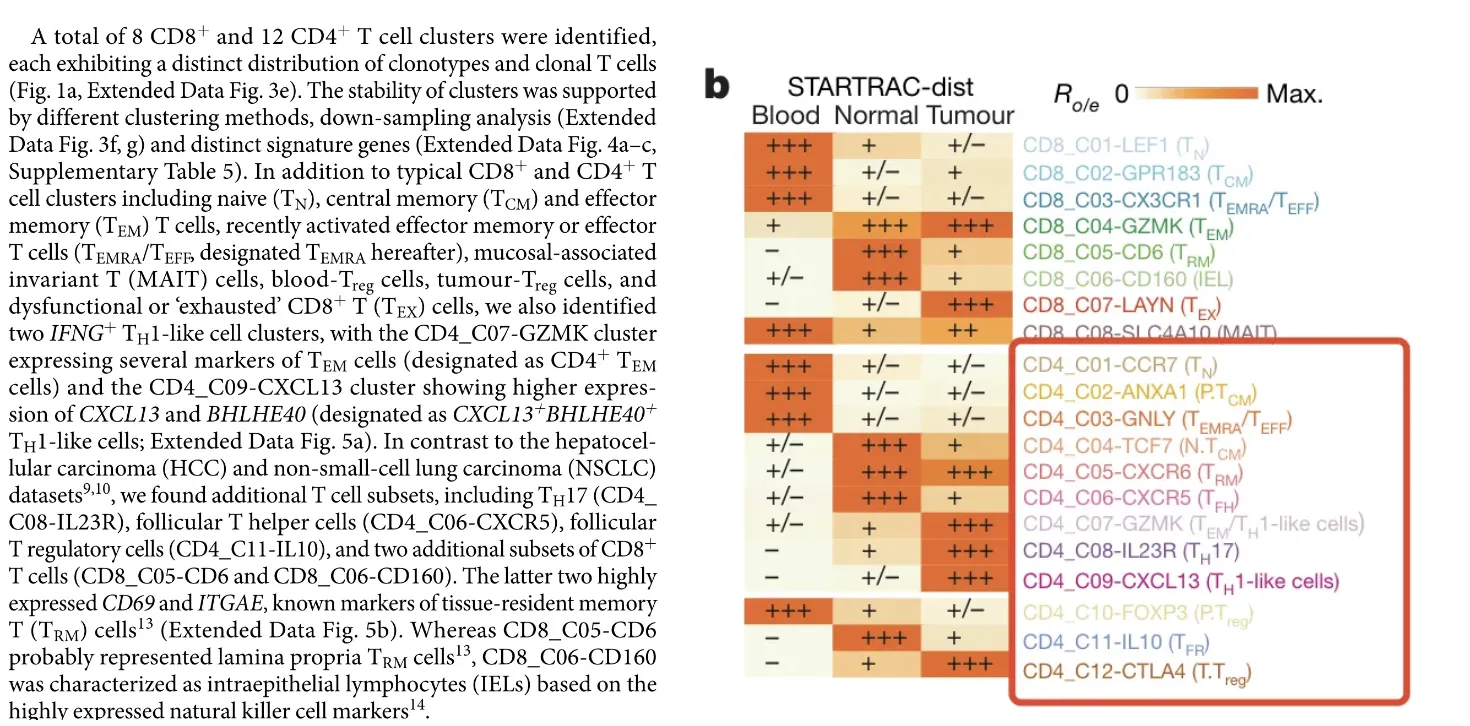
名称和标志物如下:Naive CD4+ T 细胞(CCR7);中央记忆T细胞,Tcm(ANXA1,TCF7);效应记忆/效应T细胞,Temra/Teff(GNLY);Treg细胞(FOXP3, CTLA4);Th1细胞(IFNG,CXCL13);Tem/Th1细胞(IFNG,GZMK,BHLHE40);Th17细胞(IL23R);Tfh细胞(CXCR5);组织驻留记忆T细胞,Trm(CXCR6);固有层Trm细胞,P.Trm(CD6);
CD4_markers_step3 =list(
T_check=c("CD3D","CD3E","CD4",'CD8A',"PTPRC"),
Naive=c("CCR7"),
Tcm=c("ANXA1","TCF7"),
`Temra/Teff`=c("GNLY"),
Treg=c("FOXP3","CTLA4"),
Th1 = c("IFNG","CXCL13"),
`Th1/Th1`=c("GZMK","BHLHE40"),
Th17=c("IL23R"),
Tfh=c("CXCR5"),
Trm=c("CXCR6"),
P.Trm = c("CD6")
)
genes_to_check = lapply(CD4_markers_step3, function(x) str_to_upper(x))
DotPlot(sce, features = genes_to_check ) +
# coord_flip() +
theme(axis.text.x=element_text(angle=45,hjust = 1))
w=length( unique(unlist(genes_to_check)) )/5+12;w
ggsave(filename = "CD4_T_markers_step3.pdf", width = w)
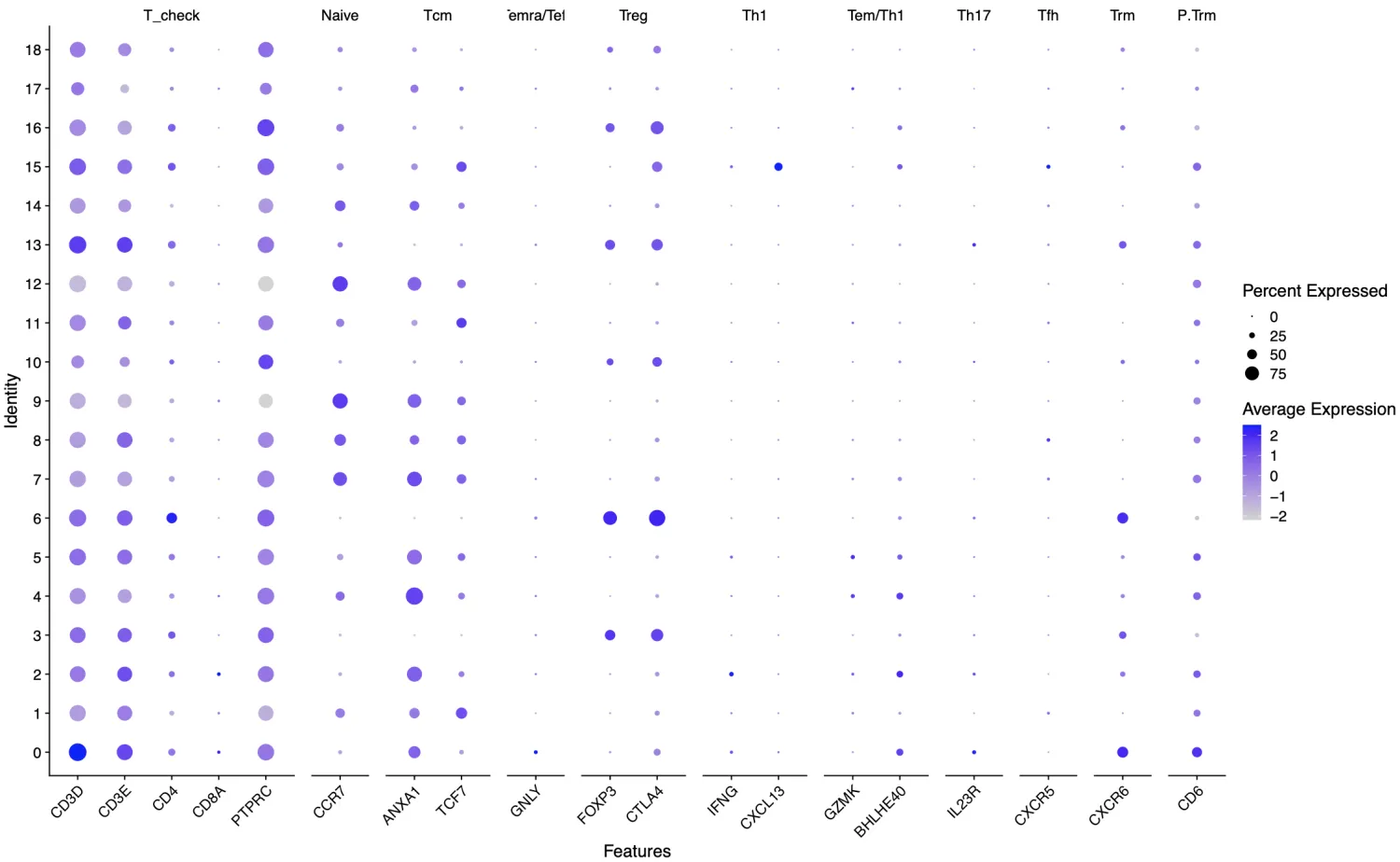
结合上面的结果对剩下的第0,1,2,4,5,7,8,9,11,12,14,15,17簇进行注释,其中第7-9,12簇可能为Naive CD4+细胞,第0,1,2,4,5,11,14簇可能是记忆性T细胞(Tcm),第5簇可能是Th1细胞。但是说实话,根据上面的结果笔者没有办法明确的认定这些细胞簇分别代表什么细胞。
3.基于转录因子分析结果注释
TFs在单细胞数据中能够较有效地区分细胞亚群,主要因为它们决定细胞命运并调控基因表达,不同细胞类型往往具有特异性的 TFs 表达模式。此外,TFs 通过调控基因网络(GRN)影响细胞的分子特征,并在细胞分化轨迹中起关键作用,揭示不同阶段的细胞变化。因此,通过分析 TFs 的表达模式和调控关系,可以精准识别细胞亚群并解析其功能特性。
3.1 导入
rm(list = ls())
library(qs)
library(Seurat)
sce <- qread("./9-CD4+T/sub_data_cd4+T.qs")
dir.create("10-pyscenic")
setwd("10-pyscenic")
3.2 数据预处理
library(Seurat)
library(SCopeLoomR)
# 将sce对象中的数据转置并保存为CSV
counts <- GetAssayData(sce, slot = "counts")
# 保存为loom文件
library(loomR)
loom <- create(filename = "input.loom",
data = counts, overwrite = TRUE)
loom$close()
rm(loom)
setwd("..")
后续都会用这个input.loom文件数据,如果loom经常出现被锁定的情况就用python语言去转换。
3.3 pyscenic分析
pyscenic环境安装,在终端/linux中运行,适用于Mac。其中需要用到的文件都在网盘里面。详细内容可至参考资料中阅读既往推文。
CONDA_SUBDIR=osx-64 conda create -n pyscenic_3.7 python=3.7
conda activate pyscenic_3.7
pip install pyscenic
conda install numpy
conda install pandas
conda activate pyscenic_3.7
#GRN-step1
pyscenic grn \
--num_workers 10 \
--output step1out_grn.tsv \
--method grnboost2 \
input.loom \
hsa_hgnc_tfs.motifs-v10.txt
#GRN-step2
pyscenic ctx \
step1out_grn.tsv \
hg38_500bp_up_100bp_down_full_tx_v10_clust.genes_vs_motifs.rankings.feather \
hg38_10kbp_up_10kbp_down_full_tx_v10_clust.genes_vs_motifs.rankings.feather \
--annotations_fname motifs-v10nr_clust-nr.hgnc-m0.001-o0.0.tbl \
--expression_mtx_fname input.loom \
--output step2out_ctx.tsv \
--mode "dask_multiprocessing" \
--num_workers 10 \
--mask_dropouts
#GRN-step3
pyscenic aucell \
input.loom \
step2out_ctx.tsv \
--output out_SCENIC.loom \
--num_workers 10
3.4 pyscenic下游分析
rm(list = ls())
library(stringr)
library(Seurat)
library(SCopeLoomR)
library(AUCell)
library(SCENIC)
library(dplyr)
library(KernSmooth)
library(RColorBrewer)
library(plotly)
library(BiocParallel)
library(grid)
library(ComplexHeatmap)
library(data.table)
library(SCENIC)
library(qs)
library(BiocParallel)
register(MulticoreParam(workers = 8, progressbar = TRUE))
loom <- open_loom("./10-pyscenic/out_SCENIC.loom")
sce <- qread("./9-CD4+T/sub_data_cd4+T.qs")
dir.create("./10-pyscenic")
setwd("./10-pyscenic")
3.5 数据预处理
regulons_incidMat <- get_regulons(loom, column.attr.name="Regulons")
regulons_incidMat[1:4,1:4]
# 将regulons_incidMat转换成一个含有不同regulon的列表
regulons <- regulonsToGeneLists(regulons_incidMat)
class(regulons)
# 在loom文件中提取名称为RegulonsAUC的信息
regulonAUC <- get_regulons_AUC(loom,column.attr.name='RegulonsAUC')
head(regulonAUC)[1:3,1:3]
# 提取regulon的阈值
regulonAucThresholds <- get_regulon_thresholds(loom)
tail(regulonAucThresholds[order(as.numeric(names(regulonAucThresholds)))])
# 提取loom文件的嵌入信息(事实上按照之前的pyscenic分析时没有坐标信息的)
embeddings <- get_embeddings(loom)
embeddings
close_loom(loom)
rownames(regulonAUC)
names(regulons)
3.6 导入seurat对象和加载的regulon信息进行匹配应对
# 取交集
sub_regulonAUC <- regulonAUC[,match(colnames(sce),colnames(regulonAUC))]
dim(sub_regulonAUC)
sce
#确认是否一致
identical(colnames(sub_regulonAUC), colnames(sce))
# 构建细胞类型注释信息
cellTypes <- data.frame(row.names = colnames(sce),
celltype = sce$RNA_snn_res.2)
head(cellTypes)
sub_regulonAUC[1:4,1:4]
save(sub_regulonAUC,
cellTypes,
sce,
file = 'for_rss_and_visual.Rdata')
table(sce$RNA_snn_res.2)
# 根据自己需要的信息进行划分
selectedResolution <- "celltype"
cellsPerGroup <- split(rownames(cellTypes),cellTypes[,selectedResolution])
# 保留唯一/非重复的 regulon
sub_regulonAUC <- sub_regulonAUC[onlyNonDuplicatedExtended(rownames(sub_regulonAUC)),]
dim(sub_regulonAUC)
3.7 scenic自带的方式-计算regulon特异性分数(RSS)
# regulon特异性分数(Regulon Specificity Score, RSS)
selectedResolution <- "celltype"
rss <- calcRSS(AUC=getAUC(sub_regulonAUC),
cellAnnotation=cellTypes[colnames(sub_regulonAUC),selectedResolution])
rss=na.omit(rss)
# 可以进行排序
rss <- rss[,c("0","1","2","3","4","5","6","7","8","9","10","11","12","13","14","15","16","17","18")]
rssPlot <- plotRSS(rss,
labelsToDiscard = NULL, # 指定需要在热图中排除的行或列标签
zThreshold = 2, # 设定调控子的阈值,默认1
cluster_columns = FALSE, # 是否对列进行聚类
order_rows = T, # 是否对行进行排序
thr = 0.1, # 阈值参数,用于过滤 RSS 值。默认0.01
varName = "cellType",
col.low = '#330066',
col.mid = '#66CC66',
col.high= '#FFCC33',
revCol = F,
verbose = TRUE
)
rssPlot
p <- plotly::ggplotly(rssPlot$plot)
p <- p %>%
layout(
title = "RSS Plot",
xaxis = list(title = "Celltypes"),
yaxis = list(title = "Regulons")
)
p
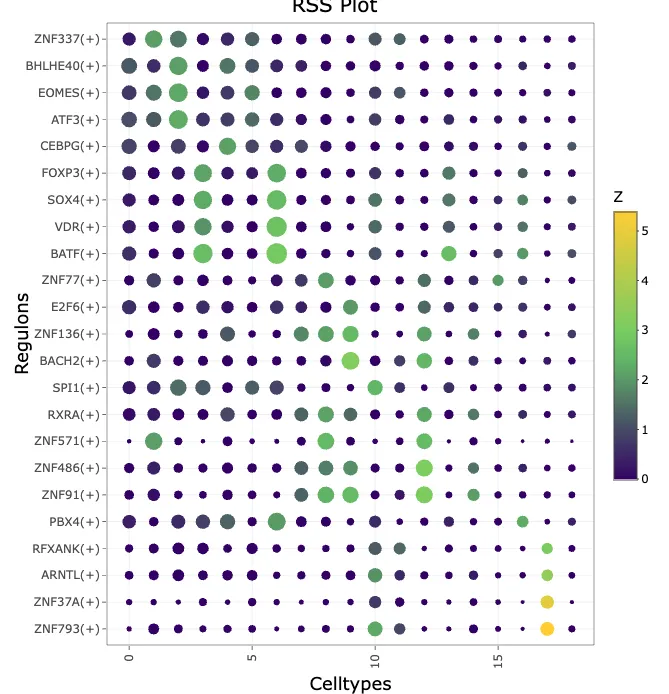
3.8 计算TFs平均活性
# 计算每个细胞组中各调控子(regulon)的平均活性,并将这些平均活性值存储在一个矩阵中
# cellsPerGroup这里得到是不同细胞群中的样本列表
# function(x)rowMeans(getAUC(sub_regulonAUC)[,x])可以计算每个细胞群的regulon平均AUC值
regulonActivity_byGroup <- sapply(cellsPerGroup,
function(x)
rowMeans(getAUC(sub_regulonAUC)[,x]))
range(regulonActivity_byGroup)
# 过滤
regulonActivity_byGroup_filtered <- regulonActivity_byGroup[apply(regulonActivity_byGroup, 1, function(row) all(row >= 0.05)), ]
regulonActivity_byGroup <- regulonActivity_byGroup_filtered
# 对结果进行归一化
regulonActivity_byGroup_Scaled <- t(scale(t(regulonActivity_byGroup),
center = T, scale=T))
# 同一个regulon在不同cluster的scale处理
dim(regulonActivity_byGroup_Scaled)
regulonActivity_byGroup_Scaled=regulonActivity_byGroup_Scaled[]
regulonActivity_byGroup_Scaled=na.omit(regulonActivity_byGroup_Scaled)
3.9 可视化-展示转录因子平均活性(全部)
library(ComplexHeatmap)
library(circlize)
Heatmap(
regulonActivity_byGroup_Scaled,
name = "z-score",
col = colorRamp2(seq(from=-2,to=2,length=11),
rev(brewer.pal(11, "Spectral"))),
show_row_names = TRUE,
show_column_names = TRUE,
row_names_gp = gpar(fontsize = 12),
clustering_method_rows = "ward.D2",
clustering_method_columns = "ward.D2",
row_title_rot = 0,
cluster_rows = TRUE,
cluster_row_slices = FALSE,
cluster_columns = FALSE
)
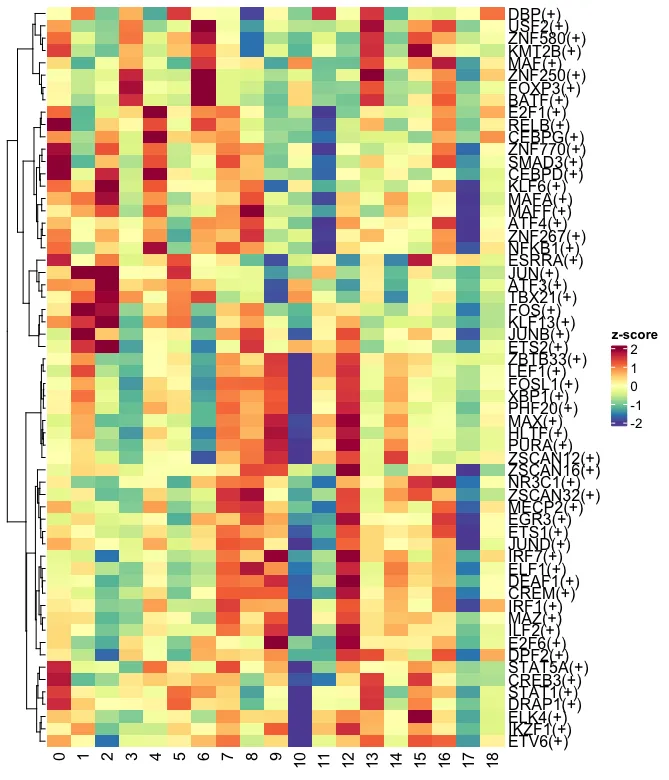
3.10 基于平均活性
这种方式是采用了将得到的scaled Data进行不同组别的差异分析
library(dplyr)
rss=regulonActivity_byGroup_Scaled
head(rss)
df = do.call(rbind,
lapply(1:ncol(rss), function(i){
dat= data.frame(
path = rownames(rss), # 当前regulon的名称
cluster = colnames(rss)[i], # 当前cluster的名称
sd.1 = rss[,i], # 当前cluster中每个调控因子的值
sd.2 = apply(rss[,-i, drop = FALSE], 1, median) #除了当前cluster之外的所有cluster 中该调控因子的中位值
)
}))
df$fc = df$sd.1 - df$sd.2
top5 <- df %>%
group_by(cluster) %>%
top_n(5, fc)
rowcn = data.frame(path = top5$cluster)
n = rss[top5$path,]
rownames(rowcn) = rownames(n)
breaksList = seq(-1.5, 1.5, by = 0.1)
colors <- colorRampPalette(c("#336699", "white", "tomato"))(length(breaksList))
pdf("TFs_output.pdf", width = 6, height = 30)
pheatmap(n,
annotation_row = rowcn,
color = colors,
cluster_rows = F,
cluster_cols = FALSE,
show_rownames = T,
#gaps_col = cumsum(table(annCol$Type)), # 使用排序后的列分割点
#gaps_row = cumsum(table(annRow$Methods)), # 行分割
fontsize_row = 12,
fontsize_col = 12,
annotation_names_row = FALSE)
dev.off()

综合三次转录因子活性分析结果来对细胞簇进行分类判定(也可仅选定一种),首先需明确不同细胞亚群中已知的关键转录因子:
-
Naive CD4+T 的特征性转录因子包括:Lck、ZAP70、LKLF(KLF2)等
-
Th1细胞的特征性转录因子包括:Tbet(TBX21)、STAT1、STAT4等
-
Th2细胞的特征性转录因子包括:GATA3、BATF、IRF4、STAT6等
-
Th17细胞的特征性转录因子包括:RORγt(RORC)、AHR、BATF、c-MAF(CMIP)、IκBζ、IRF4、RORα等
-
Tfh细胞的特征性转录因子包括:BATF、BCL6、c-MAF、IRF4、STAT3等
-
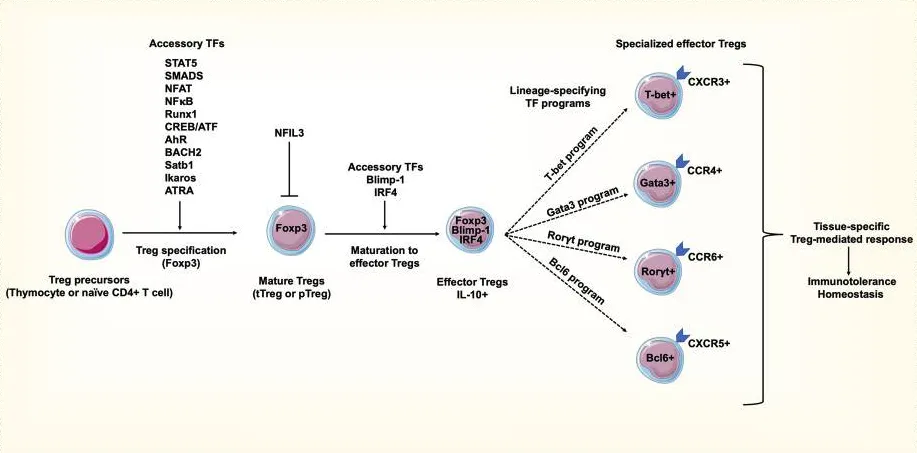
Treg细胞的特征性转录因子包括:FOXP3、STAT5、Helios(IKZF2)等
接下来用Treg细胞作为例子,既往在按照生物学背景分析时第3,6,10,13,16,18簇被认为可能为Treg细胞。Treg 细胞中最重要的转录因子是 Foxp3,此外,还有多个辅助和谱系特异性转录因子共同维持其调节性表型,包括 AHR、BACH2、BCL-11B、IKAROS、IRF4、BLIMP1、BATF、NFIL3、T-bet、GATA3、RORγt 和 BCL-6等 (PMID:37336954)。综合三次转录因子活性分析的结果其中第3、6、13、16是Treg细胞的可能性最大,均含有较高的FOXP3+。
3.11基于特定转录因子绘图
此外还可以针对FOXP3/STAT5A/IKZF2进行绘图
library(SummarizedExperiment)
library(patchwork)
seurat.data <- sce
sce$RNA_snn_res.2
Idents(seurat.data) <- "RNA_snn_res.2"
regulonsToPlot = c("FOXP3(+)","STAT5A(+)","IKZF2(+)")
regulonsToPlot %in% row.names(sub_regulonAUC)
seurat.data@meta.data = cbind(seurat.data@meta.data ,
t(assay(sub_regulonAUC[regulonsToPlot,])))
# Vis
p1 = DotPlot(seurat.data, features = unique(regulonsToPlot)) + RotatedAxis()
p2 = RidgePlot(seurat.data, features = regulonsToPlot , ncol = 2)
p3 = VlnPlot(seurat.data, features = regulonsToPlot,pt.size = 0)
p4 = FeaturePlot(seurat.data,features = regulonsToPlot)
wrap_plots(p1,p2,p3,p4)
从结果来看进一步证实了第3,6,13,16簇是Treg细胞的可能性最大(热图和特定转录因子绘图综合判断)。

接下来,按照同样的流程判断其他细胞簇的转录因子情况并同步比对cosg差异分析的结果(结果来源于上一篇推文内容)。
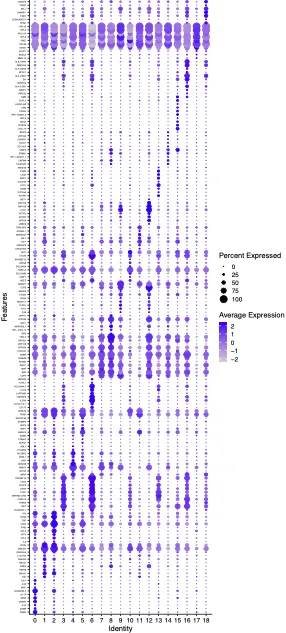
综合以上所有结果:
-
第0簇可能属于Th17细胞,因其高表达IL22和 IL17A。
-
第1、2 簇可能为Th1细胞,因其特征性标志物包括TBX21(+)。
-
第4、5、11簇可能为记忆性T细胞,因其表达ANXA1、GIMAP4、LEF1、TCF7。
-
第7-9、12、18簇可能为Naïve CD4⁺ T细胞,因其标志物为CCR7、SELL。
-
第10簇可归类为Treg细胞,因为其高表达FOXP3、CTLA4、IKZF2。
-
第14簇缺乏特异性转录因子和标志物,但ZSCAN12 表达较高,因此命名为ZSCAN12⁺T细胞。
-
第15簇也缺乏明显特异的转录因子和标志物,其中 ELK4 表达较高,因此命名为ELK4⁺T细胞。
-
第17簇同样没有显著特异的转录因子和标志物,但 ZNF793表达较高,因此命名为ZNF793⁺T细胞。
4.细胞注释
#####细胞生物学命名
celltype <- data.frame(
ClusterID = c(0:18),
celltype = c(0:18)
)
# 这里强烈依赖于生物学背景,看dotplot的基因表达量情况来人工审查单细胞亚群名字
celltype[celltype$ClusterID %in% c(7:9,12,18 ),2]='Naive T'
celltype[celltype$ClusterID %in% c(1,2 ),2]='Th1'
celltype[celltype$ClusterID %in% c(0 ),2]='Th17'
celltype[celltype$ClusterID %in% c(4,5,11),2]='Tm' # 记忆性
celltype[celltype$ClusterID %in% c(3,6,10,13,16 ),2]='Treg'
celltype[celltype$ClusterID %in% c(14 ),2]='ZSCAN12+T'
celltype[celltype$ClusterID %in% c(15 ),2]='ELK4+T'
celltype[celltype$ClusterID %in% c(17 ),2]='ZNF793+T'
sub_data <- sce
table(celltype$celltype)
table(sub_data@meta.data$RNA_snn_res.2)
table(celltype$celltype)
# ELK4+T Naive T Th1 Th17 Tm Treg ZNF793+T ZSCAN12+T
# 204 1459 818 421 1045 1528 189 248
sub_data@meta.data$celltype = "NA"
# 记得修改RNA_snn_res.0.6
for(i in 1:nrow(celltype)){
sub_data@meta.data[which(sub_data@meta.data$RNA_snn_res.2 == celltype$ClusterID[i]),'celltype'] <- celltype$celltype[i]}
table(sub_data@meta.data$celltype)
# umap/tsne
source('~/Desktop/practiceGSE188711/scRNA_scripts/Bottom_left_axis.R')
result <- left_axes(sub_data)
axes <- result$axes
label <- result$label
th=theme(axis.text.x = element_text(angle = 45,
vjust = 0.5, hjust=0.5))
library(patchwork)
celltype =DimPlot(sub_data, reduction = "umap",cols = my36colors,pt.size = 0.8,
group.by = "celltype",label = F,label.box = F) +
NoAxes() +
theme(aspect.ratio = 1) +
geom_line(data = axes,
aes(x = x,y = y,group = group),
arrow = arrow(length = unit(0.1, "inches"),
ends="last", type="closed")) +
geom_text(data = label,
aes(x = x,y = y,angle = angle,label = lab),fontface = 'italic')+
theme(plot.title = element_blank())
celltype
ggsave('umap_sub.pdf',width = 9,height = 7)
以下就是最终以及原文中的分群结果(原文中把Treg细胞去除了)。
本次分析完成了CD4⁺ T 细胞亚群注释的实践流程。事实上,在分析之前,笔者已预料到结果可能与最终结论存在较大差异,这种差异不仅源于分析技术上的细微差别,更与对CD4⁺ T 细胞的整体认知相关。尽管本次分群结果未必是最优方案,但笔者认为至少不能算是错误,整个流程仍具备实操学习的价值。此外,如果针对肿瘤细胞进行注释,可能还需要增加富集分析,以提高注释的准确性和生物学解释的深度。
参考资料
-
CD4+ T cells in cancer; Nat Cancer. 2023 Mar;4(3):317-329
-
OMIP-030: Characterization of human T cell subsets via surface markers. Cytometry A. 2015 Dec;87(12):1067-9.
-
Tumour heterogeneity and intercellular networks of nasopharyngeal carcinoma at single cell resolution. Nat Commun. 2021 Feb 2;12(1):741.
-
Liver tumour immune microenvironment subtypes and neutrophil heterogeneity. Nature. 2022 Dec;612(7938):141-147.
-
Immune phenotypic linkage between colorectal cancer and liver metastasis. Cancer Cell. 2022 Apr 11;40(4):424-437.e5.
-
Transcriptional and epigenetic basis of Treg cell development and function: its genetic anomalies or variations in autoimmune diseases. Cell Res. 2020 Jun;30(6):465-474.
-
The role of transcription factors in shaping regulatory T cell identity. Nat Rev Immunol. 2023 Dec;23(12):842-856.
-
生信技能树:https://mp.weixin.qq.com/s/cehCf99Z4pA5n_o63NmXGg https://mp.weixin.qq.com/s/JU1g7VOHekzv7XmncCZ62g https://mp.weixin.qq.com/s/AehBIahuY9Fy_sQcShx-Eg
-
生信方舟:https://mp.weixin.qq.com/s/sL_8YFulHsZ42L8G5DyY8w https://mp.weixin.qq.com/s/Y6Z3tjGVj-7unTirRak5tA
-
芒果师兄:https://mp.weixin.qq.com/s/muXTFCp0oqSFDcuUc4mItA
-
生信人:https://mp.weixin.qq.com/s/kk1YkbdbPi8VGgYXGzPhUw
-
赛默飞-中国官网:免疫学工作资源中心界面
-
流式中文网:https://www.sohu.com/a/219918954_610701
致谢:感谢曾老师以及生信技能树团队全体成员。更多精彩内容可关注公众号:生信技能树,单细胞天地,生信菜鸟团等公众号。
注:若对内容有疑惑或者有发现明确错误的朋友,请联系后台(欢迎交流)。更多相关内容可关注公众号:生信方舟

















 1730
1730

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









