







该推文首发于公众号:单细胞天地
在上一讲中完成了inferCNV的分析流程,本讲将回顾学习单细胞分析中的三个工具:Ro/e、Augur、miloR,这三个工具分别能够判断细胞簇在组织分布倾向、扰动影响和细胞差异丰度。
本次内容涉及到的工程文件可通过网盘获得:中级篇2,链接: https://pan.baidu.com/s/1y-HHLXoXsJbgWKCdz26-gQ 提取码: yx93 。
此外,可以向“生信技能树”公众号发送关键词‘单细胞’,直接获取Seurat V5版本的完整代码。
Ro/e——判断细胞簇在组织分布倾向的分析工具
Ro/e分析又称为STARTRAC-distribution(张泽民院士团队开发的STARTRAC框架中的其中一个工具)的主要目的是通过计算组织中观测细胞数与期望细胞数(期望细胞数通过卡方检验计算得出)的比率。

与卡方检验中定义的 (观测值-期望细胞数)²/期望细胞数仅能反映观测值与期望细胞数的偏离程度不同,STARTRAC通过Ro/e 可进一步指示特定细胞簇在特定组织中是否富集或耗竭。如下图中是分析不同的T细胞簇/巨噬细胞簇在不同组织中的Ro/e富集情况。
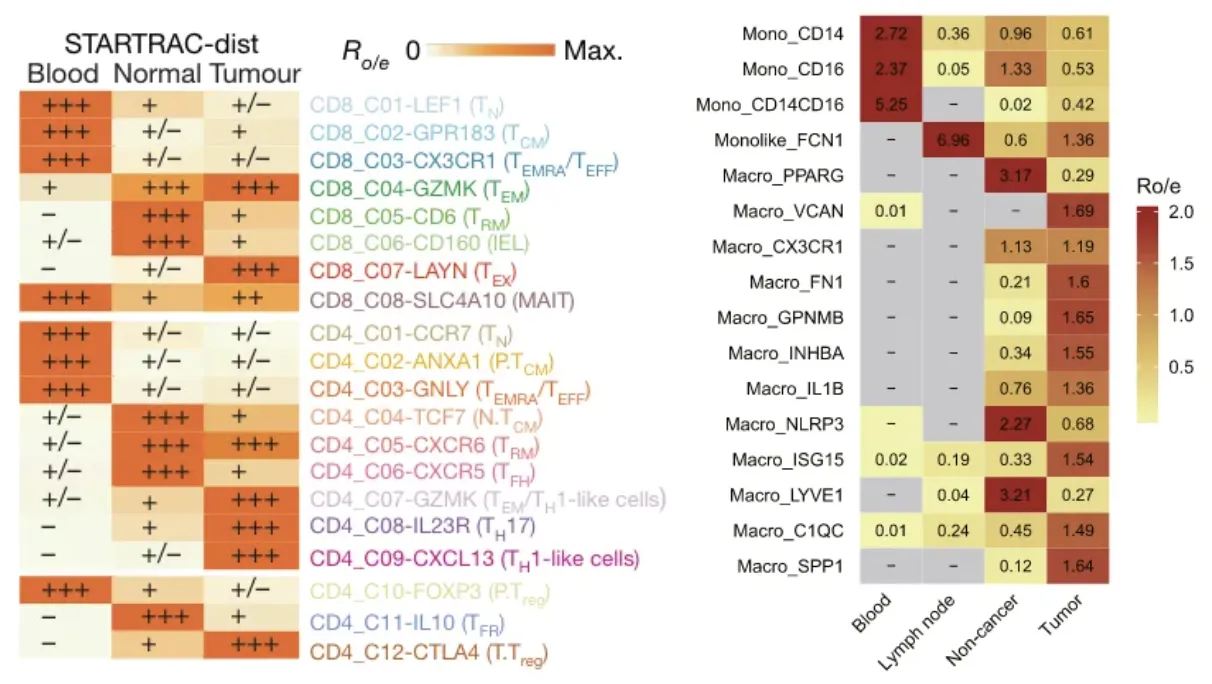
Ro/e 指数的评分规则 :
如果 Ro/e>1,则表明在该组织中,某细胞簇的细胞数量高于期望细胞数,即表现为富集;如果 Ro/e<1,则表明某细胞簇在该组织中的细胞数量低于期望细胞数,即表现为消耗。 +++, Ro/e > 1; ++, 0.8 < Ro/e ≤ 1; +, 0.2 ≤ Ro/e ≤ 0.8; +/−, 0 < Ro/e < 0.2; −, Ro/e = 0

个人认为期望细胞数也是基于现有数据进行测算的并且最后生成的值是比率,所以其实最终结果跟细胞数本身很有关系,同时细胞数量也不应太少,容易出现很大的偏倚。
Ro/e 指数分析步骤
1.导入
rm(list = ls())
library(Startrac)
library(ggplot2)
library(tictoc)
library(ggpubr)
library(ComplexHeatmap)
library(RColorBrewer)
library(circlize)
library(tidyverse)
library(sscVis)
library(Seurat)
library(tidyverse)
library(readr)
library(qs)
library(BiocParallel)
register(MulticoreParam(workers = 8, progressbar = TRUE))
scRNA <- qread("./9-CD4+T/CD4+t_final.qs")
Idents(scRNA) <- "celltype"
DimPlot(scRNA)
2.数据预处理
data <- scRNA@meta.data
colnames(data)
# [1] "orig.ident" "nCount_RNA" "nFeature_RNA" "percent_mito"
# [5] "percent_ribo" "percent_hb" "RNA_snn_res.0.01" "seurat_clusters"
# [9] "RNA_snn_res.0.05" "RNA_snn_res.0.1" "RNA_snn_res.0.2" "RNA_snn_res.0.3"
# [13] "RNA_snn_res.0.5" "RNA_snn_res.0.8" "RNA_snn_res.1" "celltype"
# [17] "location" "pANN_0.25" "DF.classifications" "contamination"
# [21] "RNA_snn_res.0.4" "RNA_snn_res.0.6" "RNA_snn_res.0.7" "RNA_snn_res.0.9"
# [25] "RNA_snn_res.1.1" "RNA_snn_res.1.2" "RNA_snn_res.1.3" "RNA_snn_res.1.4"
# [29] "RNA_snn_res.1.5" "RNA_snn_res.1.6" "RNA_snn_res.1.7" "RNA_snn_res.1.8"
# [33] "RNA_snn_res.1.9" "RNA_snn_res.2"
# 需要得到clone.id(ID标识),patient(样本),majorCluster(细胞亚群),loc(不同组织/位置)列
# 并且需要重新命名
data <- data[,c(34,1,16,17)]
colnames(data) <- c("clone.id","patient","majorCluster","loc")
data$Cell_Name <- c("T cell")
data$patient <- as.character(data$patient)
data$clone.id <- as.character(data$clone.id)
data$majorCluster <- as.character(data$majorCluster)
data$loc <- as.character(data$loc)
3.Ro/e分析
Roe <- calTissueDist(data,
byPatient = F,
colname.cluster = "majorCluster", # 不同细胞亚群
colname.patient = "patient", # 不同样本
colname.tissue = "loc", # 不同组织
method = "chisq", # "chisq", "fisher", and "freq"
min.rowSum = 0)
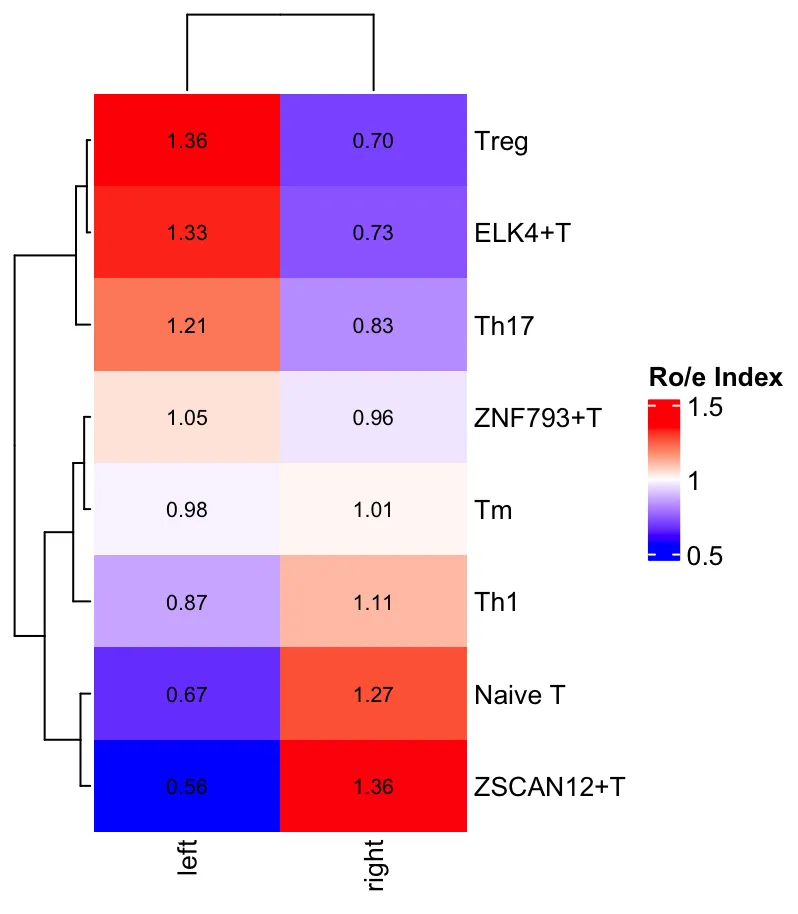
Roe
# left right
# ELK4+T 1.3286764 0.7309727
# Naive T 0.6669732 1.2725882
# Th1 0.8664169 1.1093401
# Th17 1.2137669 0.8250281
# Tm 0.9822346 1.0145413
# Treg 1.3638576 0.7021762
# ZNF793+T 1.0462073 0.9621785
# ZSCAN12+T 0.5643888 1.3565553
# 这是最关键的值,然后进行可视化即可
4.可视化
table(data$majorCluster)
# ELK4+T Naive T Th1 Th17 Tm Treg ZNF793+T ZSCAN12+T
# 204 1459 818 421 1045 1528 189 248
col_fun <- colorRamp2(c(min(Roe, na.rm = TRUE), 1, max(Roe, na.rm = TRUE)),
c("blue", "white", "red"))
Heatmap(as.matrix(Roe),
show_heatmap_legend = TRUE,
cluster_rows = TRUE,
cluster_columns = TRUE,
row_names_side = 'right',
show_column_names = TRUE,
show_row_names = TRUE,
col = col_fun,
row_names_gp = gpar(fontsize = 10),
column_names_gp = gpar(fontsize = 10),
heatmap_legend_param = list(
title = "Ro/e Index", # 自定义图注名称
at = seq(0.5, 1.5, by = 0.5), # 例刻度的位置/自己的数据必须修改一下!
labels = seq(0.5, 1.5, by = 0.5) # 每个刻度的标签/自己的数据必须修改一下!
),
cell_fun = function(j, i, x, y, width, height, fill) {
grid.text(sprintf("%.2f", Roe[i, j]), x, y, gp = gpar(fontsize = 8, col = "black"))
}
)
# cell_fun = function(j, i, x, y, width, height, fill)
# j 和 i:分别表示单元格的列和行索引。
# x 和 y:是单元格的中心位置坐标,用于确定文本的位置。
# width 和 height:表示单元格的宽度和高度,可以用来调整文本位置或大小。
# fill:表示单元格的背景颜色。
setwd("..")
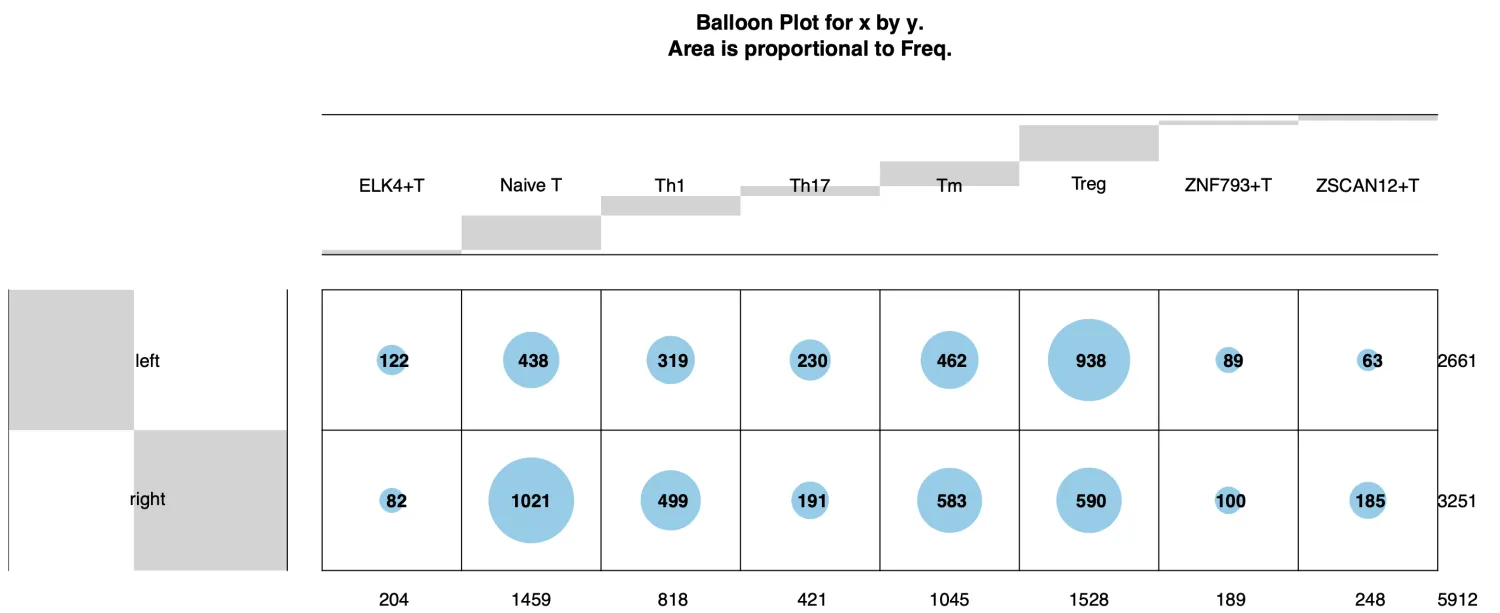
从细胞数和Ro/e值可以看到左半结肠中存在较多数量的Treg细胞以及较高的Ro/e值,Naive T细胞则在右半结肠中更多和显著的Ro/e值。因此,这也符合现有的生物学知识,右半结肠癌通常具有更高的免疫原性而左半结肠癌则相反。
Augur——判断扰动影响的分析工具
Augur能够在单细胞数据中优先考虑对生物扰动最敏感的细胞类型,其采用机器学习框架来量化扰动和未扰动细胞在高维空间中的可分性(比如不同的疾病状态,药物刺激等)。Augur 使用的机器学习框架依赖于分类算法,如随机森林(Random Forests)或逻辑回归(Logistic Regression),这些算法非常适合处理具有高维特征空间的单细胞数据。
Augur的具体做法是:通过判断是否能够仅凭分子特征准确预测出每个细胞的实验处理标签(如处理组 vs 对照组),来量化这种可分离性。在实际操作中,它会为每种细胞类型单独训练一个机器学习模型,用于预测每个细胞来自哪个实验条件。接着,通过交叉验证评估每个细胞类型特定分类器的准确性,从而为细胞类型的优先级排序提供一个定量的依据。
Augur分析流程
1.导入
rm(list = ls())
library(stringr)
library(Seurat)
library(Augur)
library(dplyr)
library(patchwork)
library(viridis)
library(qs)
library(BiocParallel)
register(MulticoreParam(workers = 8, progressbar = TRUE))
scRNA <- qread("./9-CD4+T/CD4+t_final.qs")
Idents(scRNA) <- "celltype"
DimPlot(scRNA)
dir.create("./12-STARTRAC-miloR-Augur")
setwd("./12-STARTRAC-miloR-Augur")
2.单一组别不同条件下augur分析
比如治疗前后,但是本次分析就把左右半结肠位置数据当做实验分组了。
# 此函数的主要目的是评估并计算每种细胞类型在特定条件(如实验组别或时间点)下的表现,通常通过计算区域下曲线(AUC)来量化。它是单一条件下的细胞类型表现的度量,可以帮助识别在特定实验设置下哪些细胞类型表现最为显著或重要。
augur <- calculate_auc(scRNA,
cell_type_col = "celltype", # 细胞亚群数据
label_col = "location", # 实验分组数据
n_threads = 8)
qsave(augur,"augur.qs")
head(augur$AUC, 5)
# # A tibble: 5 × 2
# cell_type auc
# <chr> <dbl>
# 1 Naive T 0.628
# 2 Th17 0.621
# 3 Th1 0.604
# 4 ZSCAN12+T 0.588
# 5 ELK4+T 0.586
3.可视化
# 明确哪个细胞群体的RNA水平变化最大
# plot_lollipop函数是基于ggplot框架的,所以很容易就可以美化图片
# 还可以提取原始函数进行修改
augur <- qread("augur.qs")
library(ggplot2)
plot_lollipop(augur) +
geom_segment(aes(xend = cell_type, yend = 0.5), size = 1) +
geom_point(size = 3, aes(color = cell_type)) + # 增加点的大小和颜色映射
scale_color_manual(values = c(
"Naive T" = "#1f77b4", # 蓝色
"Th17" = "#ff7f0e", # 橙色
"Th1" = "#2ca02c", # 绿色
"ZSCAN12+T" = "#d62728", # 红色
"ELK4+T" = "#9467bd", # 紫色
"Treg" = "#8c564b", # 棕色
"Tm" = "#e377c2", # 粉色
"ZNF793+T" = "#7f7f7f" # 灰色
))
# 结合umap
# rank模式
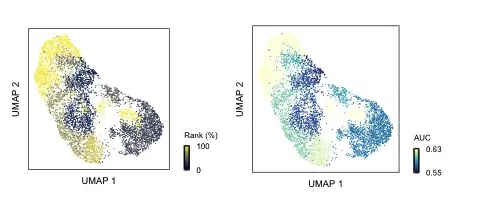
plot_umap(augur,scRNA,
mode = "rank",
reduction = "umap",
palette = "cividis",
cell_type_col = "celltype")
# default模式
plot_umap(augur,scRNA,
mode = "default",
reduction = "umap",
palette = "YlGnBu", # "viridis", "plasma", "magma", "inferno"
cell_type_col = "celltype")
setwd("..")
plot_lollipop展示了在不同处理情况下(group)中变化最显著的细胞(AUC值),从结果来看Naive T细胞受到的扰动程度是最大的,ZNF793+T收到的扰动程度则是最小的。
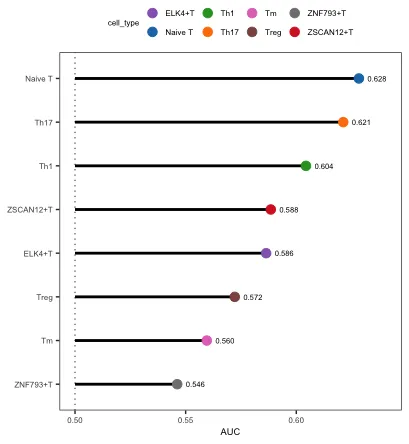
mode中的rank和default模式的区别在于是否用AUC值进行颜色映射。
miloR——判断细胞差异丰度的分析工具
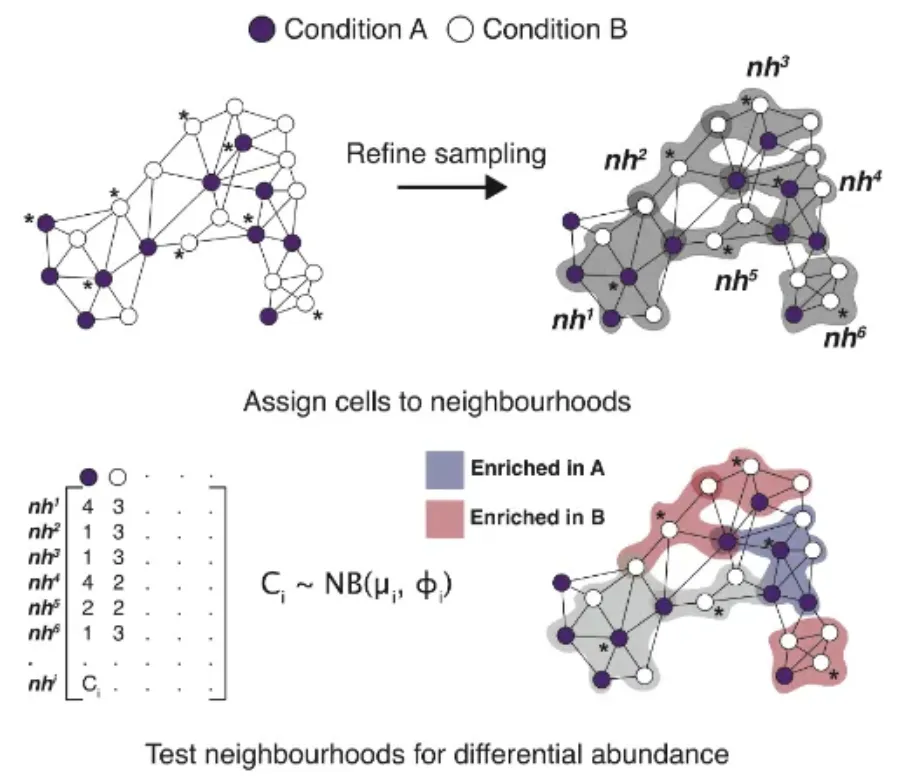
miloR能够随机定义细胞中的细胞节点,然后通过K最近邻法去识别所定义节点与周围的其他细胞节点之间的邻近关系,找到跟这些节点最近的(比如欧几里得法)其他细胞节点,将这些具有邻近关系的细胞小群看做不同的群组,此时可以想象一下,每个群组中都会存在具有疾病和正常信息的许多细胞,接着再将这些群组中疾病与否的细胞数分别进行比较和汇总,最后可以得到基于某一大类细胞中内部细胞变化趋势的结果(比如是否倾向与疾病的方向)。
有了这个结果之后,结合预先定义的细胞分群结果有助于使用者更好的去了解疾病与否、疾病状态、药物刺激等情况下的不同细胞亚群的变化状态,因此使用miloR可以有效的作为单细胞水平上细胞差异分析的补充工具。
miloR分析流程
1.导入
rm(list = ls())
library(stringr)
library(Seurat)
library(miloR)
library(scater)
library(scran)
library(dplyr)
library(patchwork)
library(qs)
library(BiocParallel)
register(MulticoreParam(workers = 8, progressbar = TRUE))
scRNA <- qread("./9-CD4+T/CD4+t_final.qs")
Idents(scRNA) <- "celltype"
DimPlot(scRNA)
dir.create("./12-STARTRAC-miloR-Augur")
setwd("./12-STARTRAC-miloR-Augur")
2.数据预处理
需要一个seurat对象,建议提前预处理
# check
table(scRNA$orig.ident)
# GSM5688706_WGC GSM5688707_JCA GSM5688708_LS-CRC3 GSM5688709_RS-CRC1 GSM5688710_R_CRC3
# 738 542 1381 1879 807
# GSM5688711_R_CRC4
# 565
table(scRNA$celltype)
# ELK4+T Naive T Th1 Th17 Tm Treg ZNF793+T ZSCAN12+T
# 204 1459 818 421 1045 1528 189 248
3.构建miloR对象
# 由于需要输入SingleCellExperiment对象
# 因此先进行转化一下
scRNA_pre <- as.SingleCellExperiment(scRNA)
# 构建miloR对象
scRNA_milo <- Milo(scRNA_pre)
reducedDim(scRNA_milo,"UMAP") <- reducedDim(scRNA_pre,"UMAP")
scRNA_milo
# class: Milo
# dim: 21308 5912
# metadata(0):
# assays(3): counts logcounts scaledata
# rownames(21308): RP11-34P13.7 FO538757.2 ... GRIK1-AS1 AP001626.2
# rowData names(0):
# colnames(5912): GSM5688706_WGC_AACACACGTATCACGT-1 GSM5688706_WGC_AACACACTCAAGGTGG-1 ...
# GSM5688711_R_CRC4_TTTGGTTTCTGGGATT-1 GSM5688711_R_CRC4_TTTGTTGGTAATGCTC-1
# colData names(35): orig.ident nCount_RNA ... RNA_snn_res.2 ident
# reducedDimNames(3): PCA HARMONY UMAP
# mainExpName: RNA
# altExpNames(0):
# nhoods dimensions(2): 1 1
# nhoodCounts dimensions(2): 1 1
# nhoodDistances dimension(1): 0
# graph names(0):
# nhoodIndex names(1): 0
# nhoodExpression dimension(2): 1 1
# nhoodReducedDim names(0):
# nhoodGraph names(0):
# nhoodAdjacency dimension(2): 1 1
4.构建K最邻图谱
# d值与降维时选定的pca值一致
# k值与FindNeighbors时选定的值一致
scRNA_milo <- buildGraph(scRNA_milo, k = 30, d = 15,reduced.dim = "PCA")
5.在 KNN 图上定义具有代表性的邻域
# 官网建议prop在0.1-0.2
# K和d和KNN图参数一致
scRNA_milo <- makeNhoods(scRNA_milo, prop = 0.2, k = 30, d = 15,
refined = TRUE, reduced_dims = "PCA")
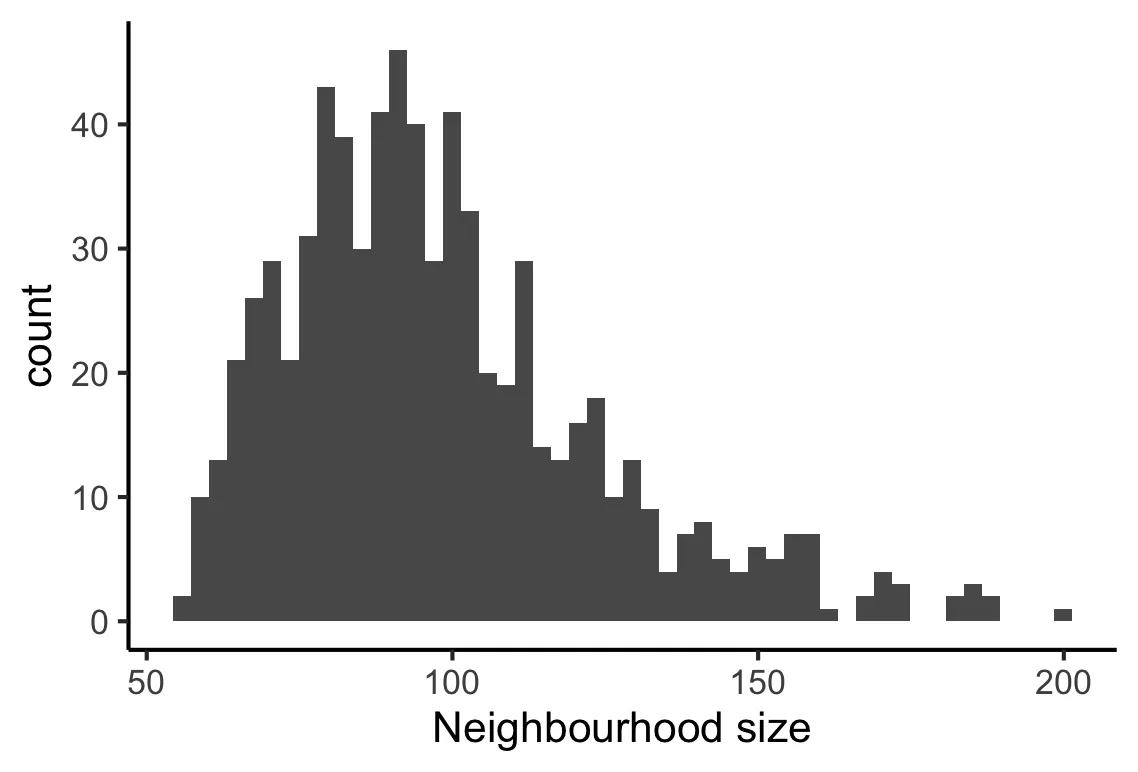
plotNhoodSizeHist(scRNA_milo)

开发者建议在50到100达到峰值,否则需要增加k/prop的值。
6.计算neighbourhoods中的细胞
计算每个样本中有多少细胞位于每个邻域中。
scRNA_milo <- countCells(scRNA_milo,
meta.data = as.data.frame(colData(scRNA_milo)),
sample="orig.ident") # sample代表样本
# colData(scRNA) 相当于代表了metadata的信息
# sample代表了样本数
head(nhoodCounts(scRNA_milo))
# 这个步骤能够给scRNA数据中增加一个n*m的matrix
# n代表不同的neighborhood数量,m代表实验样本
# 6 x 6 sparse Matrix of class "dgCMatrix"
# GSM5688706_WGC GSM5688707_JCA GSM5688708_LS-CRC3 GSM5688709_RS-CRC1 GSM5688710_R_CRC3 GSM5688711_R_CRC4
# 1 19 7 41 18 . .
# 2 1 . 5 3 2 61
# 3 5 2 84 3 . .
# 4 10 21 4 30 . .
# 5 21 14 59 15 . .
# 6 71 . 19 4 . 3
这为对象添加了一个矩阵,其中n是邻域的数量,𝑚是实验样本的数量。值表示每个样本在邻域中计数的细胞数量。这个计数矩阵将用于差异丰度测试。

7.实验设计定义

研究者把在使用文件中把condition定义为covariate(协变量),然而在正式分组的时候又把condition定义为实验分组,个人觉得协变量是定义错了,condition就是实验分组。本次数据中location对标condition。
traj_design <- data.frame(colData(scRNA_milo))[,c("orig.ident", "location")]
traj_design <- distinct(traj_design)
rownames(traj_design) <- traj_design$orig.ident
#重新排序行名以匹配nhoodCounts(milo)的列
traj_design <- traj_design[colnames(nhoodCounts(scRNA_milo)), , drop=FALSE]
traj_design$location <- factor(traj_design$location, level = c("left", "right"))
traj_design
# orig.ident location
# GSM5688706_WGC GSM5688706_WGC left
# GSM5688707_JCA GSM5688707_JCA left
# GSM5688708_LS-CRC3 GSM5688708_LS-CRC3 left
# GSM5688709_RS-CRC1 GSM5688709_RS-CRC1 right
# GSM5688710_R_CRC3 GSM5688710_R_CRC3 right
# GSM5688711_R_CRC4 GSM5688711_R_CRC4 right
8.计算邻里连通性
Milo使用cydar引入的Spatial FDR校正的变体,该校正考虑了邻域之间的重叠。具体来说,每个假设检验的P值都通过k个最近邻距离的倒数进行加权。要使用这个统计量,首先需要在Milo对象中存储最近邻之间的距离。
# Milo使用cydar引入的Spatial FDR校正的变体,该校正考虑了邻域之间的重叠。具体来说,每个假设检验的P值都通过k个最近邻距离的倒数进行加权。要使用这个统计量,首先需要在Milo对象中存储最近邻之间的距离。
scRNA_milo <- calcNhoodDistance(scRNA_milo, d=15, reduced.dim = "PCA")
9.检测结果
正式测试,明确定义实验设计,这里使用了location
# 把批次和分组信息加到design中去
da_results <- testNhoods(scRNA_milo,design = ~ location, design.df = traj_design)
da_results %>%
arrange(SpatialFDR) %>%
head()
# logFC logCPM F PValue FDR Nhood SpatialFDR
# 15 -8.035409 10.53289 7.723075 0.005486977 0.1453135 15 0.1009631
# 25 7.663408 10.18627 8.654687 0.003287987 0.1453135 25 0.1009631
# 31 8.492067 10.93321 8.159168 0.004314811 0.1453135 31 0.1009631
# 131 -8.311036 10.77975 7.523350 0.006127759 0.1453135 131 0.1009631
# 183 8.511668 10.94935 8.097942 0.004462580 0.1453135 183 0.1009631
# 231 -8.242297 10.71764 7.567548 0.005979667 0.1453135 231 0.1009631
# 可以看一下柱状图和点图
ggplot(da_results, aes(PValue)) + geom_histogram(bins=50)
ggplot(da_results, aes(logFC, -log10(SpatialFDR))) +
geom_point() +
geom_hline(yintercept = 1) ## Mark significance threshold (10% FDR)
10.可视化
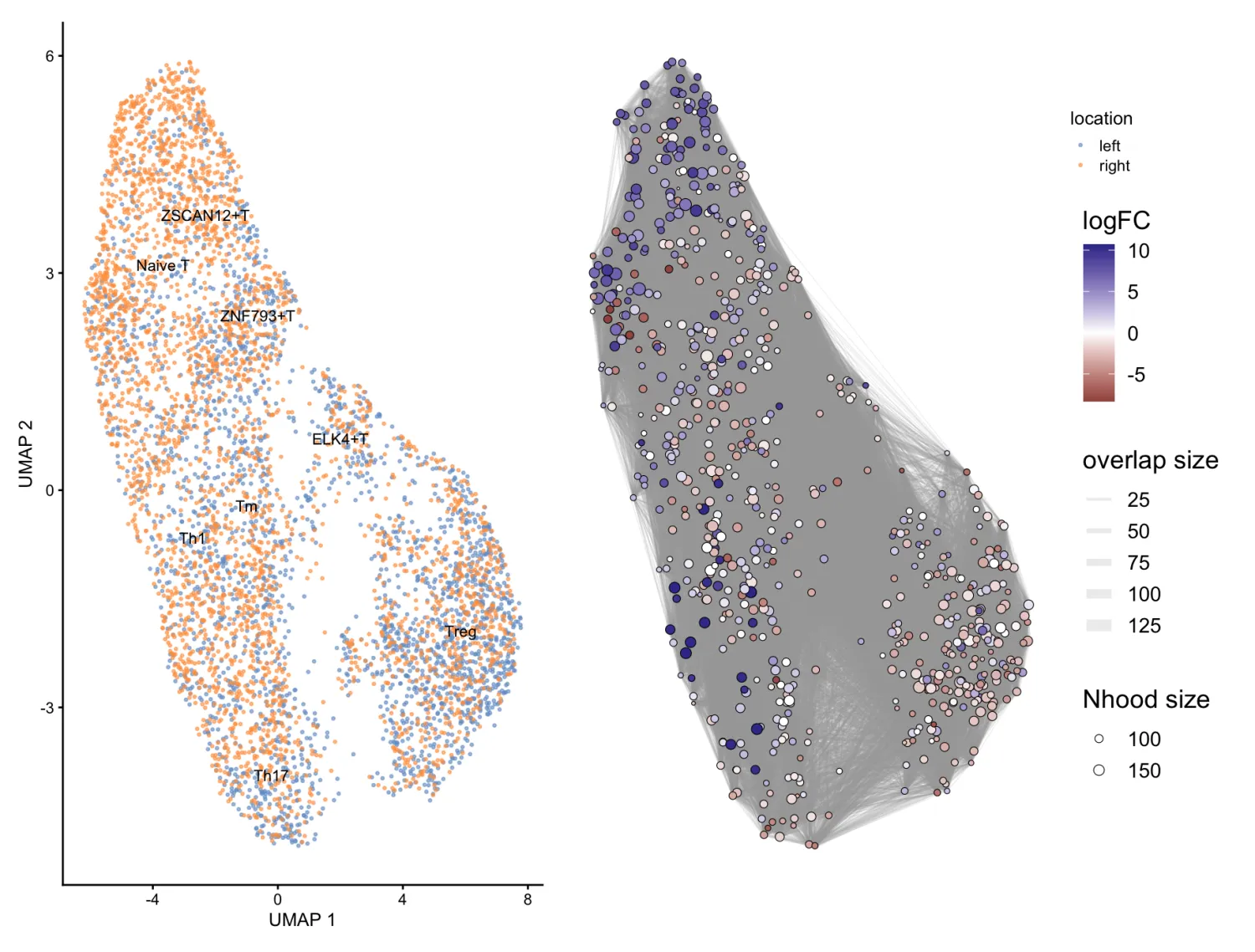
scRNA_milo <- buildNhoodGraph(scRNA_milo)
## Plot single-cell UMAP
umap_pl <- plotReducedDim(scRNA_milo, dimred = "UMAP",
colour_by="location", text_by = "celltype",
text_size = 3, point_size=0.5) + guides(fill="none")
umap_pl
## Plot neighbourhood graph
nh_graph_pl <- plotNhoodGraphDA(scRNA_milo, da_results,
layout="UMAP",alpha=0.9) #alpha默认0.1
umap_pl + nh_graph_pl +
plot_layout(guides="collect")
UMAP 图(左图):显示了不同细胞类型在两个实验组之间的分布。图中使用了两种颜色,蓝色(left) 和 橙色(right),分别代表left组和right组。不同的细胞类型被明确标注,并且可以看到left组和right组之间的细胞分布。
邻域网络图(右图):右边的图是基于MiloR分析的邻域图。每个圈代表一个邻域,邻域的大小与其中细胞的数量成比例。颜色梯度表示了logFC值:正的 logFC(蓝色)意味着该邻域中的细胞在right组中富集,负的 logFC(红色)则表明该邻域中的细胞在left组中富集。邻域之间的连线表示邻域之间的相似性或重叠,线条的厚度表示邻域之间的重叠度(overlap size),越厚的线表示两个邻域之间的细胞更为相似。
11.数据注释
da_results <- annotateNhoods(scRNA_milo,
da_results,
coldata_col = "celltype")
head(da_results)
# logFC logCPM F PValue FDR Nhood SpatialFDR celltype celltype_fraction
# 1 -1.574239 9.515303 0.4603793 0.49750211 0.6649894 1 0.6489640 Naive T 0.8596491
# 2 2.920511 10.169145 2.1800639 0.13991968 0.3845433 2 0.3461536 Naive T 0.8021978
# 3 4.925942 10.883476 4.0560689 0.04410711 0.2888600 3 0.2281237 Naive T 0.8735632
# 4 -2.618949 11.056240 1.2512551 0.26340764 0.4888901 4 0.4599119 Tm 0.8437500
# 5 -2.062491 10.447327 1.0799157 0.29880501 0.5154568 5 0.4895583 Tm 0.4098361
# 6 1.986811 10.452844 1.0385032 0.30825677 0.5173425 6 0.4939665 Th1 0.5952381
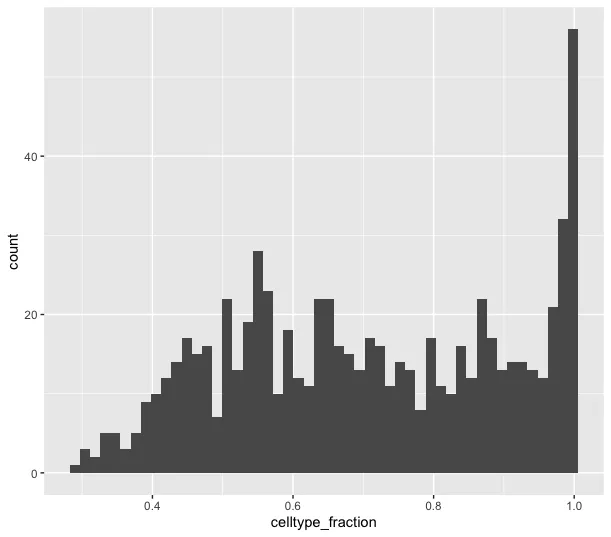
ggplot(da_results, aes(celltype_fraction)) + geom_histogram(bins=50)
str(da_results)
table(da_results$celltype)
range(da_results$SpatialFDR)
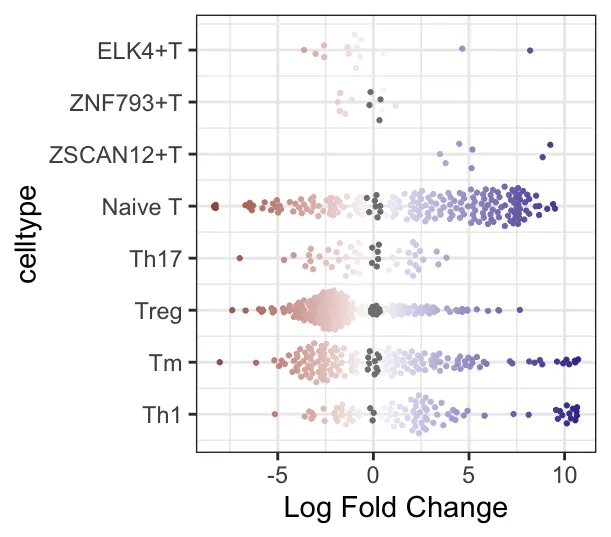
plotDAbeeswarm(da_results, group.by = "celltype",alpha = 0.9) # alpha默认0.1
celltype_fraction 直方图:展示了邻域中某种特定细胞类型的比例分布(celltype_fraction)。Y轴代表邻域的数量,X轴表示邻域中细胞类型所占的比例(范围从0到1)。如果邻域的细胞类型比例接近1,说明大多数邻域是单一细胞类型主导的,这意味着邻域中的细胞几乎都是同一种类型,没有明显的混合细胞类型。这种结果通常出现在较为纯净的细胞群或具有显著分离特征的细胞亚群。
logFC 与细胞类型关联的 beeswarm 图:展示了不同细胞类型的logFC在不同邻域中的分布。Y轴表示细胞类型,X轴表示logFC(即不同组之间在邻域中的差异丰度变化),每个点代表一个邻域。比如Naive T细胞似乎在right组中的邻域具有更大的logFC值,Treg细胞在两组中的logFC值似乎差不多,但在left组中邻域数量更多且集中在-2.5左右。
12.找到DA群体的marker
## Add log normalized count to Milo object
scRNA_milo <- logNormCounts(scRNA_milo)
# alpha默认0.1,所以SpatialFDR < 0.9是修改过的
da_results$NhoodGroup <- as.numeric(da_results$SpatialFDR < 0.9 & da_results$logFC < 0)
da_nhood_markers <- findNhoodGroupMarkers(scRNA_milo, da_results,
subset.row = rownames(scRNA_milo),
aggregate.samples = TRUE, # 建议设置为TRUE
sample_col = "orig.ident")
head(da_nhood_markers)
# GeneID logFC_1 adj.P.Val_1 logFC_0 adj.P.Val_0
# 1 A1BG 0.069556844 0.8845036 -0.069556844 0.8845036
# 2 A1BG-AS1 -0.034653314 0.8845036 0.034653314 0.8845036
# 3 A1CF 0.000000000 1.0000000 0.000000000 1.0000000
# 4 A2M 0.002280312 0.9963105 -0.002280312 0.9963105
# 5 A2M-AS1 -0.004230877 1.0000000 0.004230877 1.0000000
# 6 A2ML1 0.000000000 1.0000000 0.000000000 1.0000000
12.1.自动对邻域进行分组
#在许多情况下,DA邻域位于KNN图的不同区域,将所有重要的 DA 群体分组在一起可能并不理想,因为它们可能包括非常不同细胞类型的细胞。
# 对于这种场景,开发者用了一个邻里函数,它使用社区检测根据(1)两个邻域之间共享的小区数量将邻域划分为组;(2)DA 社区的倍数变化方向;(3)折叠变化的差异。
## Run buildNhoodGraph to store nhood adjacency matrix
scRNA_milo <- buildNhoodGraph(scRNA_milo)
## Find groups
da_results <- groupNhoods(scRNA_milo, da_results,
max.lfc.delta = 2,da.fdr = 0.9)#da.fdr默认0,1
head(da_results)
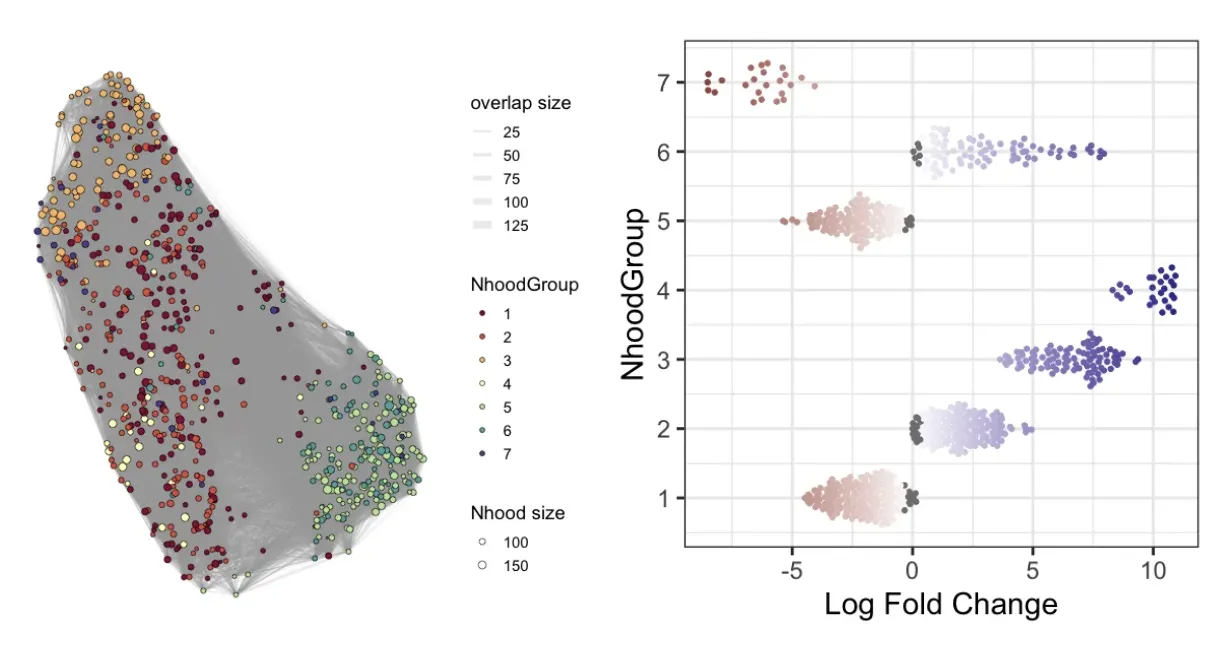
plotNhoodGroups(scRNA_milo, da_results, layout="UMAP")
plotDAbeeswarm(da_results, "NhoodGroup",alpha = 0.9) #alpha默认是0.1
这个步骤是按照DA的结果进行自动划分簇,后续步骤还可以找到差异基因。
12.2 找到特征基因
## Exclude zero counts genes
keep.rows <- rowSums(logcounts(scRNA_milo)) != 0
scRNA_milo <- scRNA_milo[keep.rows, ]
## Find HVGs
dec <- modelGeneVar(scRNA_milo)
hvgs <- getTopHVGs(dec, n=2000)
head(hvgs)
# [1] "HSPA1A" "HSPA1B" "ANXA1" "S100A4" "DNAJB1" "TXNIP"
# 找到每个邻里群的one-vs-all差异基因表达
nhood_markers <- findNhoodGroupMarkers(scRNA_milo, da_results, subset.row = hvgs,
aggregate.samples = TRUE, sample_col = "orig.ident")
head(nhood_markers)[1:4,1:4]
# GeneID logFC_1 adj.P.Val_1 logFC_2
# 1 ABCA1 0.09845269 0.4335701 -0.034954072
# 2 ABCA2 0.13412658 0.1315646 -0.047259161
# 3 ABCA5 0.01590351 0.7922435 0.010316026
# 4 ABCA7 -0.01051437 0.8831909 0.001969199
# 找到group2的差异基因
gr2_markers <- nhood_markers[c("logFC_2", "adj.P.Val_2")]
colnames(gr2_markers) <- c("logFC", "adj.P.Val")
head(gr2_markers[order(gr2_markers$adj.P.Val), ])
# 如果明确了对第二组细胞感兴趣
nhood_markers <- findNhoodGroupMarkers(scRNA_milo, da_results, subset.row = hvgs,
aggregate.samples = TRUE, sample_col = "orig.ident",
subset.groups = c("2")
)
head(nhood_markers)
# logFC_2 adj.P.Val_2 GeneID
# LARP4 0.2920211 0.0000272445 LARP4
# OAS3 0.2884843 0.0002802494 OAS3
# NFKBID 0.3886595 0.0003353456 NFKBID
# SSBP2 0.3669311 0.0007693966 SSBP2
# CD8B 0.3266638 0.0010654284 CD8B
# MRPL18 0.4196821 0.0067862948 MRPL18
# 如果明确了相比较的邻域
nhood_markers <- findNhoodGroupMarkers(scRNA_milo, da_results, subset.row = hvgs,
subset.nhoods = da_results$NhoodGroup %in% c('2','5'),
aggregate.samples = TRUE, sample_col = "orig.ident")
head(nhood_markers)
# GeneID logFC_2 adj.P.Val_2 logFC_5 adj.P.Val_5
# 1 ABCA1 0.007280498 0.63548063 -0.007280498 0.63548063
# 2 ABCA2 -0.024945154 0.40911416 0.024945154 0.40911416
# 3 ABCA5 0.021147177 0.25841475 -0.021147177 0.25841475
# 4 ABCA7 -0.015377090 0.66034055 0.015377090 0.66034055
# 5 ABCD2 -0.006936849 0.65999062 0.006936849 0.65999062
# 6 ABHD18 -0.053001633 0.04725136 0.053001633 0.04725136
12.3 差异基因可视化
# 参数自行修改
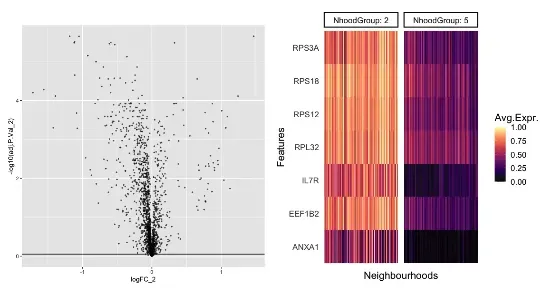
ggplot(nhood_markers, aes(logFC_2,-log10(adj.P.Val_2 ))) +
geom_point(alpha=0.5, size=0.5) +
geom_hline(yintercept = 0.05)
# 筛选标准可以自行修改
markers <- nhood_markers$GeneID[nhood_markers$adj.P.Val_2 < 0.05 & nhood_markers$logFC_2 > 1]
plotNhoodExpressionGroups(scRNA_milo, da_results, features=markers,
subset.nhoods = da_results$NhoodGroup %in% c('2','5'),
scale=TRUE,
grid.space = "fixed")
12.4 同一邻域但是不同干预的细胞比较
dge_5 <- testDiffExp(scRNA_milo, da_results, design = ~ location,
meta.data = data.frame(colData(scRNA_milo)),
subset.row = rownames(scRNA_milo),
subset.nhoods=da_results$NhoodGroup=="5")
dge_5
# $`5`
# logFC AveExpr t P.Value adj.P.Val B Nhood.Group
# RPS26 2.05155879 2.16078022 40.913548 2.133985e-259 3.832637e-255 583.478172 5
# TSC22D3 1.18443050 1.71490401 18.676109 2.678121e-71 2.404953e-67 151.256496 5
# TXNIP 0.92242005 0.78962782 17.282387 4.814448e-62 2.882250e-58 130.022537 5
# IL7R 1.06746697 1.32047401 16.570152 1.643201e-57 7.377973e-54 119.625571 5
# UGP2 0.80880323 0.85367836 16.342942 4.291897e-56 1.541649e-52 116.376331 5
# FAM118A -0.63992629 0.59464042 -15.560710 2.510389e-51 7.514431e-48 105.447340 5
# ETS1 0.69424488 1.08136205 14.602241 9.954116e-46 2.553942e-42 92.618518 5
# AIM1 0.36544366 0.22221315 13.380244 5.355135e-39 1.202228e-35 77.204983 5
# MT-CO1 -0.58424851 5.05175258 -12.970636 7.565387e-37 1.509715e-33 72.284370 5
# SMAP2 0.53193670 0.77612312 12.062342 2.816038e-32 5.057605e-29 61.829911 5
# RPS9 -0.58244288 2.86336400 -11.929160 1.249072e-31 2.039394e-28 60.350991 5
# ZNF765 0.17038370 0.06098921 11.890038 1.929662e-31 2.888060e-28 59.919222 5
需要提醒一下,其中很多参数笔者做了修改也做了注释,使用的时候需要先从默认值开始分析!
本次内容完成了这三个工具的完整实践流程。虽然它们各自侧重的分析方向不同,但在实际应用中,它们的使用场景是存在交叉的。也就是说,我们可以针对同一批数据,分别使用这三个工具从不同的视角和分析维度进行解读,最终获得更加全面、立体的生物学信息。
参考资料:
-
Lineage tracking reveals dynamic relationships of T cells in colorectal cancer. Nature. 2018 Dec;564(7735):268-272.
-
A pan-cancer single-cell transcriptional atlas of tumor infiltrating myeloid cells. Cell. 2021 Feb 4;184(3):792-809.e23.
-
Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat Med. 2018 Jul;24(7):978-985.
-
Cell type prioritization in single-cell data. Nat Biotechnol. 2021 Jan;39(1):30-34.
-
Differential abundance testing on single-cell data using k-nearest neighbor graphs. Nat Biotechnol. 2022 Feb;40(2):245-253.
-
STARTRAC github:https://github.com/Japrin/STARTRAC
-
Augur github:https://github.com/neurorestore/Augur
-
miloR github:https://rawcdn.githack.com/MarioniLab/miloR/7c7f906b94a73e62e36e095ddb3e3567b414144e/vignettes/milo_gastrulation.html#5_Finding_markers_of_DA_populations
-
单细胞天地:https://mp.weixin.qq.com/s/FlO4fN5AlcyZ81llzHE5A https://mp.weixin.qq.com/s/yYTxb_kOjeRSkzzuUoI4mA
-
生信菜鸟团:https://mp.weixin.qq.com/s/D0nGq3XGZRIlso7dKo0qkQ
致谢:感谢曾老师以及生信技能树团队全体成员。更多精彩内容可关注公众号:生信技能树,单细胞天地,生信菜鸟团等公众号。
注:若对内容有疑惑或者有发现明确错误的朋友,请联系后台(欢迎交流)。更多相关内容可关注公众号:生信方舟
- END -

















 1442
1442

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









