







该推文首发于公众号:单细胞天地
在完成了1. 基础流程、2. 三个“矫正”工具、3. “差异”与“富集”两个工具、以及 4. 两个映射工具 这四个初级部分的学习后,我们将正式进入中级篇,第一讲的主题是亚细胞分群。
严格来说,亚细胞分群其实更适合作为高级篇的内容,因为它涉及多个关键因素,包括基础生物学背景、技术流程熟练度,以及最新的研究进展。不少从事医学与生信交叉研究的老师也坦言,分群过程往往占据单细胞分析项目80%以上的分析时间,这一点也从侧面说明了它的重要性和复杂性。
然而,考虑到笔者自身的经验和技术水平有限,同时为了确保课程内容的有序推进(后续涉及到亚群的分析),经过多番思考,最终还是决定将亚细胞分群归入中级篇进行讲解。当然,这仅是笔者的一家之言,未必绝对正确。如果您有更佳的亚细胞分群流程,欢迎在推文下方留言交流。
该篇使用的工程文件通过网盘分享:初级篇2 链接: https://pan.baidu.com/s/1ETVEkJNCJSe9NU7_aLEPVA 提取码: znmb 。此外,可以向“生信技能树”公众号发送关键词‘单细胞’,直接获取Seurat V5版本的完整代码。
初级篇1关键内容回顾
-
查看标记结果顺序:由浅入深,重点关注,反复对比。查看结果的具体顺序(曾老师的标记库,也可自建标记库)为1. Last_markers_and_umap结果;2.查看需要特别关注或者last_markers_and_umap结果中不明确或者特定组织特定标记群结果;3.查看对比cosg/findallmarkers等差异基因分析工具得到的结果,时刻提醒自己要结合生物学知识。
-
细胞亚群认定流程:第一:判断标志物是否是细胞群所特有的。第二:需要考虑标志物的平均表达量高低,一般会倾向关注表达量很高的标志物。第三:若两种分属不同细胞亚群的标志物在同一个细胞簇中出现时,需要考虑是否是特殊的细胞群。 如果细胞群出现特殊情况,就需要回溯多个图表进行综合考量。比如需要回溯nFeature,nCount图判断是否异常(包括线粒体,核糖体和血红蛋白的值),也需查看COSG/FindAllmaker差异分析结果判断一下是否符合T细胞的特征,同时要查阅高分文献确认是否有过该类型细胞的报道。如果确认了上述情况都没有问题,那就这就是真实的结果。
-
再次确认亚群注释:首先是回看标志物的气泡图和UMAP图;如果确定无误,那么就要从样本的生物学角度考虑是否符合“逻辑”;如果都是正确的那就大胆的命名,如果拿不准那就退一步往这些细胞群的上一级命名。
分析步骤
原文内容提取了CD4+T细胞进行了后续的细胞分群,那么笔者这次也挑战一下提取CD4+T细胞并进行分群。
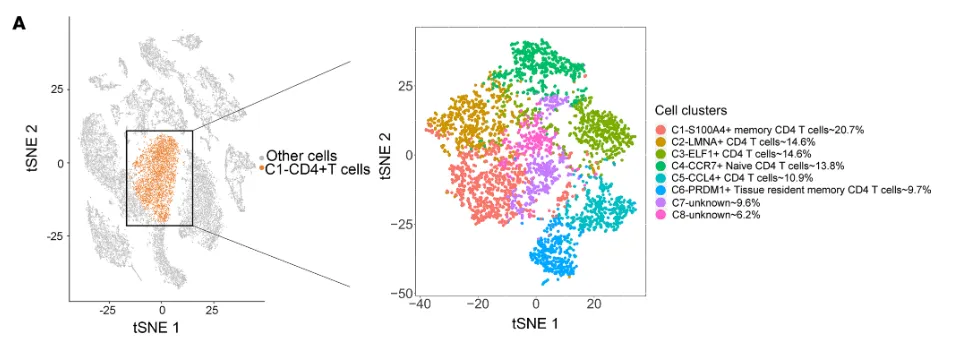
我们的分析流程主要为三步:1. 提取T/NK细胞并进行细胞“清洗”;2. 粗分T/NK细胞并进行细胞"清洗";3.提取CD4+T细胞并最终分群。
该推文实际上只完成了前两步,而这部分内容的核心就是如何合理地进行细胞“清洗”。
1.导入
rm(list=ls())
options(stringsAsFactors = F)
source('scRNA_scripts/lib.R')
source('scRNA_scripts/mycolors.R')
library(Seurat)
library(ggplot2)
library(clustree)
library(cowplot)
library(data.table)
library(dplyr)
library(qs)
library(BiocParallel)
register(MulticoreParam(workers = 8, progressbar = TRUE))
sc_dataset <- qread("./4-Corrective-data/sce.all.qs")
dir.create("./8-subcelltype/")
setwd("./8-subcelltype")
DimPlot(scRNA,label = T)
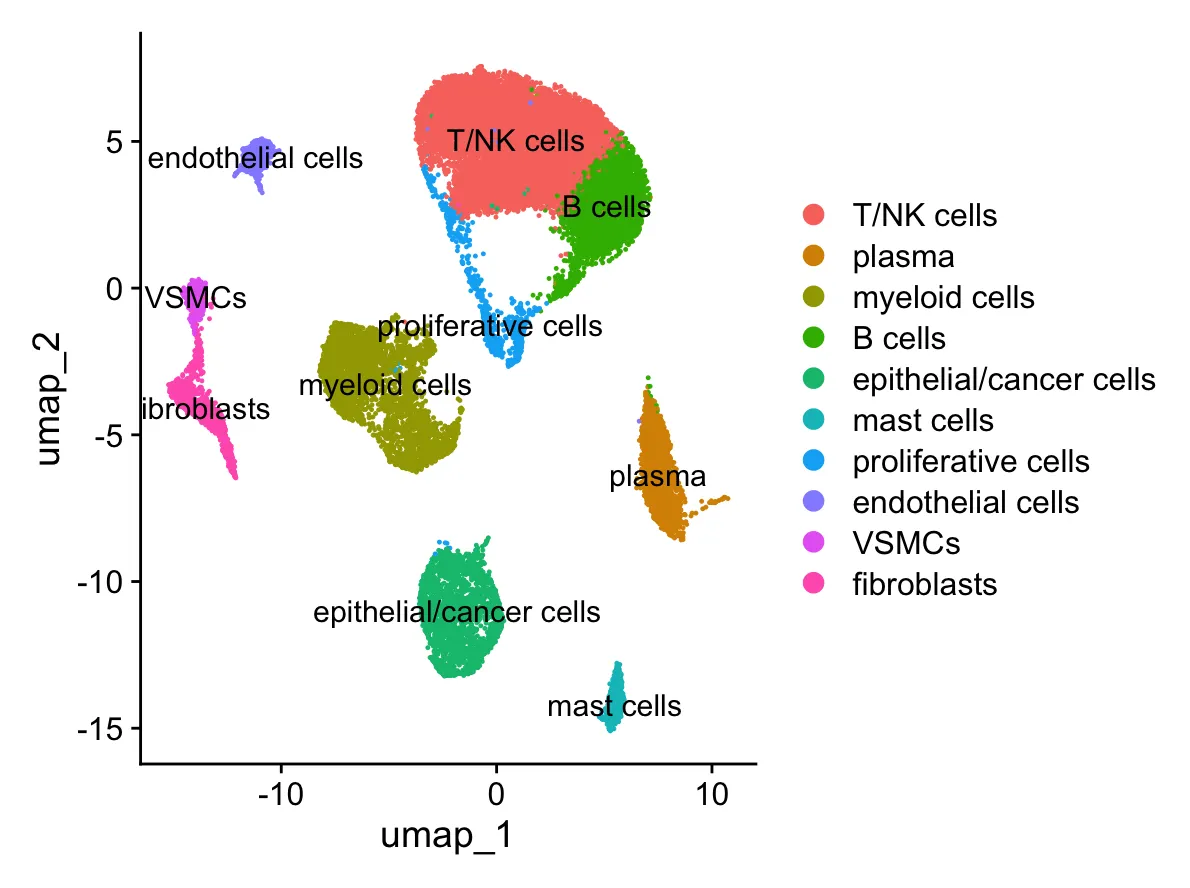
2.第一步:提取T/NK细胞并进行细胞“清洗”
2.1 提取T/NK细胞
table(sc_dataset$celltype)
Idents(sc_dataset) <- sc_dataset$celltype
sub_data <- sc_dataset[,Idents(sc_dataset) %in% c("T/NK cells")]
# check
DimPlot(sub_data,reduction = "umap",
label = TRUE, pt.size = 0.5)
观察下图,我们发现T/NK细胞聚的很紧凑,只有左下角有一个离群的细胞,说明粗分群T/NK细胞分的还不错。那么我们很多时候提取的亚群并不是如此,而且在很远的距离有很多细胞怎么办?
笔者的个人浅见:1. 首先再次确认一开始的分群是否干净(举一个不恰当的例子,假设一开始的气泡图中某一个簇中存在高表达CD3,但是同时存在弱表达的LUM和DCN,那么换句话说可能这个T细胞群中混入了成纤维细胞所以就要重新对这个簇进行再细分);2. 其次可考虑增加细胞过滤标准,比如改变min.cells和min.features的值试一试;3.再其次可以在合理的阈值范围内再适当的调试一下“矫正”工具。4.最后如果上面的流程都试过了,但还是有奇怪的细胞在,那就可以考虑人为去除。
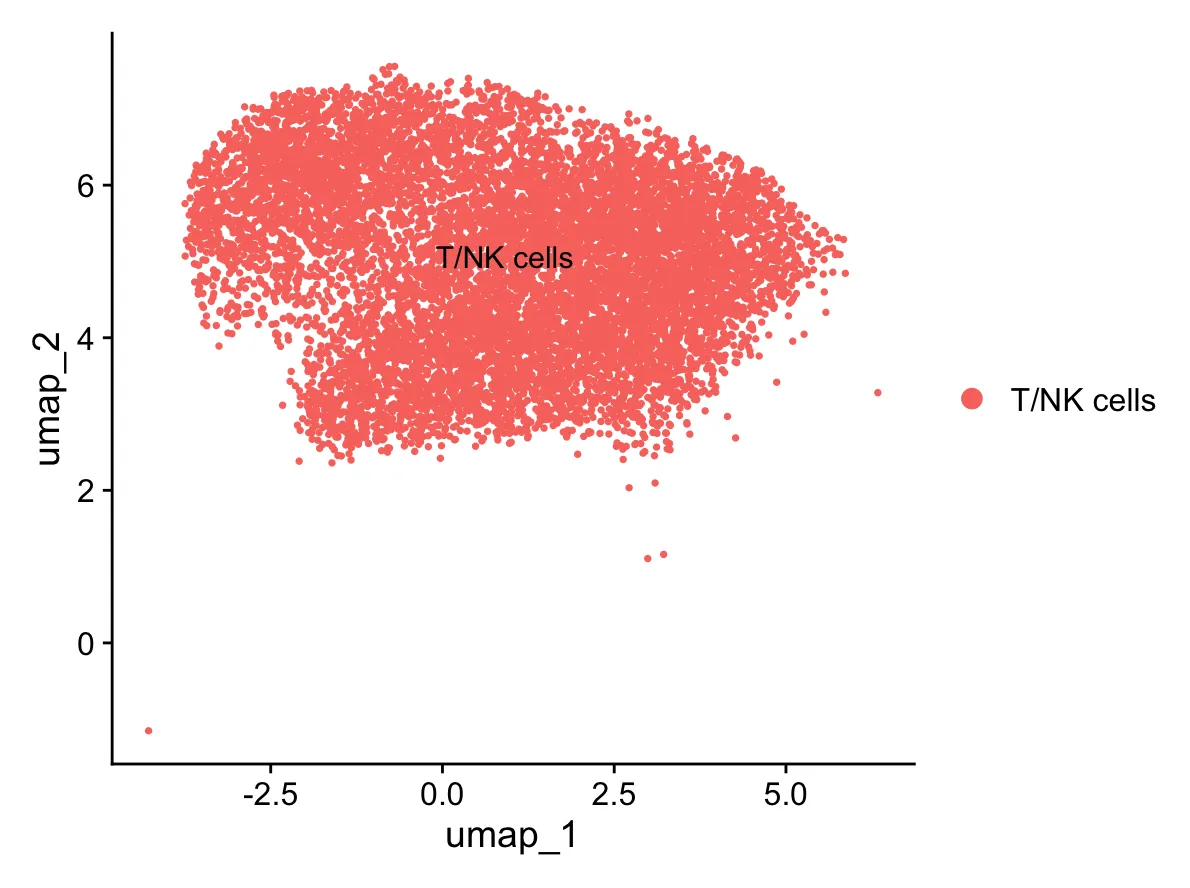
2.2.运行标准流程
标准化-找高变
sce=CreateSeuratObject(
counts = sub_data@assays$RNA$counts,
meta.data = sub_data@meta.data
)
# 标准化数据
sce <- NormalizeData(sce,
normalization.method = "LogNormalize",
scale.factor = 10000)
# 鉴定2000个高变基因(数量可人为设置,一般是2K)
sce <- FindVariableFeatures(sce,
selection.method = "vst",
nfeatures = 2000)
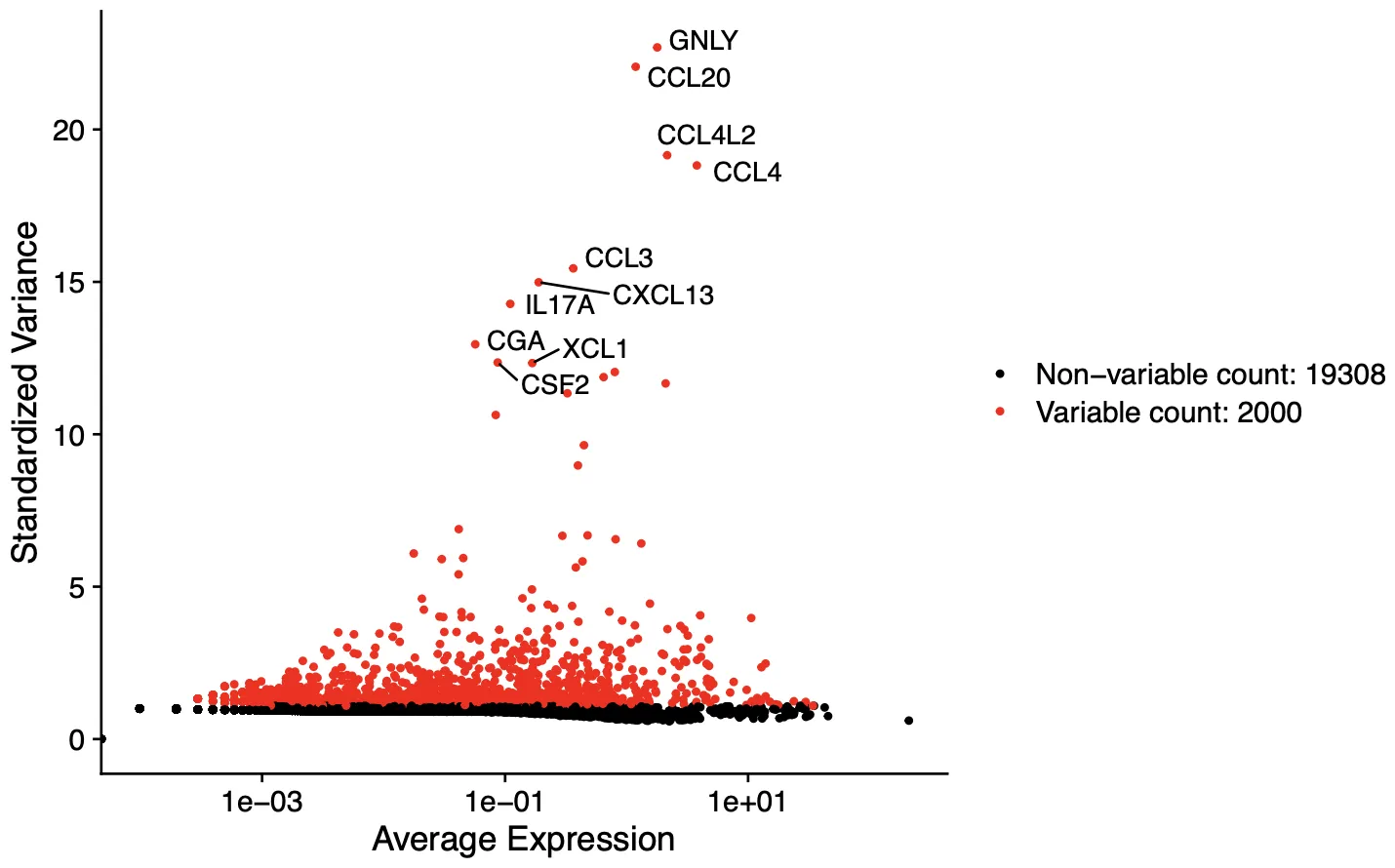
# Identify the 10 most highly variable genes
top10 <- head(VariableFeatures(sce), 10)
# 对高可变基因进行可视化
plot1 <- VariableFeaturePlot(sce)
plot2 <- LabelPoints(plot = plot1, points = top10,
repel = TRUE);plot2
ggsave("Variablegenes.pdf",plot = plot2, width = 8,height = 5,dpi = 300)
观察一下高变基因,这里的高变基因也主要与T和NK细胞相关(那就是正确的)。
T细胞亚群
-
GNLY(颗粒溶素):细胞毒性T细胞(CD8+ T细胞)和 NK细胞 的标志物,参与杀伤靶细胞。
-
IL17A:Th17细胞(辅助性T细胞亚群)的特异性标志,介导炎症反应。
-
CXCL13:滤泡辅助性T细胞(Tfh) 的标志物,促进B细胞活化和生发中心形成。
-
CCL3/CCL4/CCL20:趋化因子,常见于活化的T细胞(如Th1、Th17)或 调节性T细胞(Treg),参与招募免疫细胞。
NK细胞
-
GNLY 和 XCL1:NK细胞 的典型效应分子,参与抗病毒和抗肿瘤反应。
ScaleData+harmony整合
sce <- ScaleData(sce,features = rownames(sce))
# 正式PCA降维——————————————————————————————————
sce <- RunPCA(sce,features = VariableFeatures(object = sce) ,
#npcs = 20, # npcs = 20 表示计算并保留前20个主成分
verbose = FALSE)
# harmony整合
sce <- RunHarmony(sce,group.by.vars ="orig.ident")
#harmony降维后的可视化
DimPlot(sce, reduction = "harmony", group.by = "orig.ident")
table(sce$orig.ident)
ggsave("harmony_orig.pdf",width = 18,height = 8,dpi = 300)
# 一般也不参考,直接选择dim=15即可
#用ElbowPlot去判断PC值
ElbowPlot(sce,ndims=50)
ggsave("ElbowPlot.pdf",width = 8,height = 6,dpi = 300)
# findPC也可以参考一下
library(findPC)
stdev_sorted <- sort(sce@reductions$pca@stdev, decreasing = TRUE)
stdev_sorted
findPC(sdev = stdev_sorted, # 主成分降序排列
number = 50, # number数量不需要
method = "all", # 所有的方法都看一下,默认方法是perpendicular line
figure = T)
cluster分群
# 基于降维后的数据构建细胞邻近图
dims_N <- "15"
sce <- FindNeighbors(sce, dims = 1:dims_N,
reduction = "harmony", #单样本需要pca
verbose = FALSE)
# 进行聚类分析,基于邻近图(之前由FindNeighbors函数构建)划分细胞群体
# resolution设置了一个梯度
sce <- FindClusters(sce,
resolution = seq(0.1, 2, by = 0.1),
verbose = FALSE)
head(Idents(sce), 5)
# clustree图
library(clustree)
library(patchwork)
p1 <- clustree(sce, prefix = 'RNA_snn_res.') + coord_flip();p1
#这里的RNA_snn_res后面的数值是可以修改的
p2 <- DimPlot(sce, group.by = 'RNA_snn_res.0.6', label = T);p2
Tree_1 <- p1 + p2 + plot_layout(widths = c(3, 1));print(Tree_1)
ggsave("cluster_tree.pdf",plot = Tree_1,
width = 20,height = 12,dpi = 300)
UMAP/tSNE可视化
#确定一个想要的reslution值
sel.clust = "RNA_snn_res.0.6"
sce <- SetIdent(sce, value = sel.clust)
#Umap方式
sce <- RunUMAP(sce, dims = 1:dims_N,reduction = "harmony",
umap.method = "uwot", metric = "cosine")
UMAP_1 <- DimPlot(sce,reduction = 'umap',label = T);UMAP_1
ggsave("UMAP.pdf",plot = UMAP_1,width = 10,height = 6,dpi = 300)
#Tsne方式
# sce <- RunTSNE(sce,dims = 1:dims_N) #,reduction = "harmony")
# tsne <- DimPlot(sce,label = T,reduction = 'tsne',pt.size =1);tsne
# ggsave("TSNE.png",plot = tsne,width = 10,height = 6,dpi = 300)
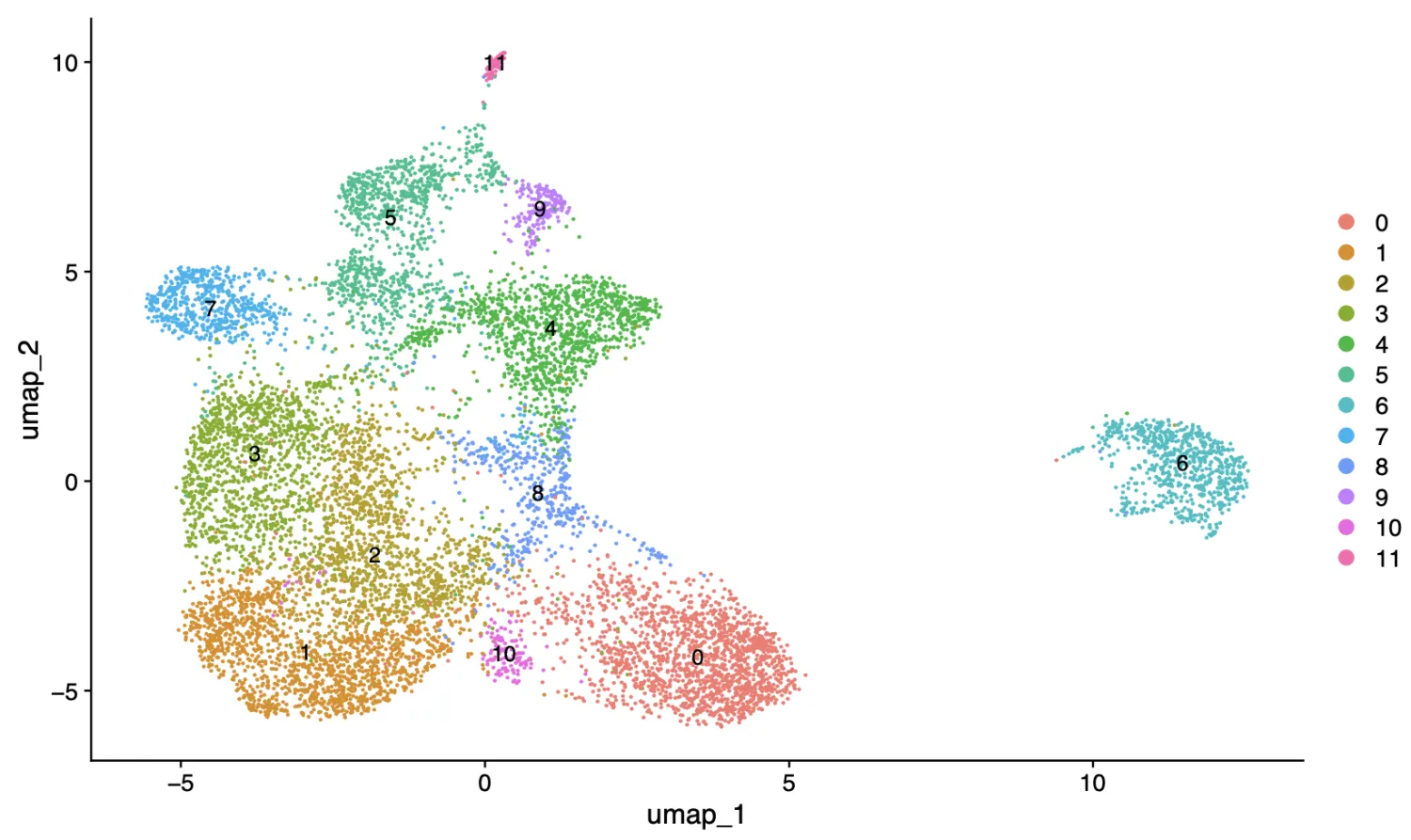
2.3.查看标记基因库(曾老师这个库真好用)
sce.all.int <- sce
sp = "human"
getwd()
dir.create('check-by-0.1')
setwd('check-by-0.1')
sel.clust = "RNA_snn_res.0.1"
sce.all.int <- SetIdent(sce.all.int, value = sel.clust)
table(sce.all.int@active.ident)
source('../../scRNA_scripts/check-all-markers.R')
setwd('../')
getwd()
dir.create('check-by-0.6')
setwd('check-by-0.6')
sel.clust = "RNA_snn_res.0.6"
sce.all.int <- SetIdent(sce.all.int, value = sel.clust)
table(sce.all.int@active.ident)
source('../../scRNA_scripts/check-all-markers.R')
setwd('../')
getwd()
dir.create('check-by-1.5')
setwd('check-by-1.5')
sel.clust = "RNA_snn_res.1.5"
sce.all.int <- SetIdent(sce.all.int, value = sel.clust)
table(sce.all.int@active.ident)
source('../../scRNA_scripts/check-all-markers.R')
setwd('../')
getwd()
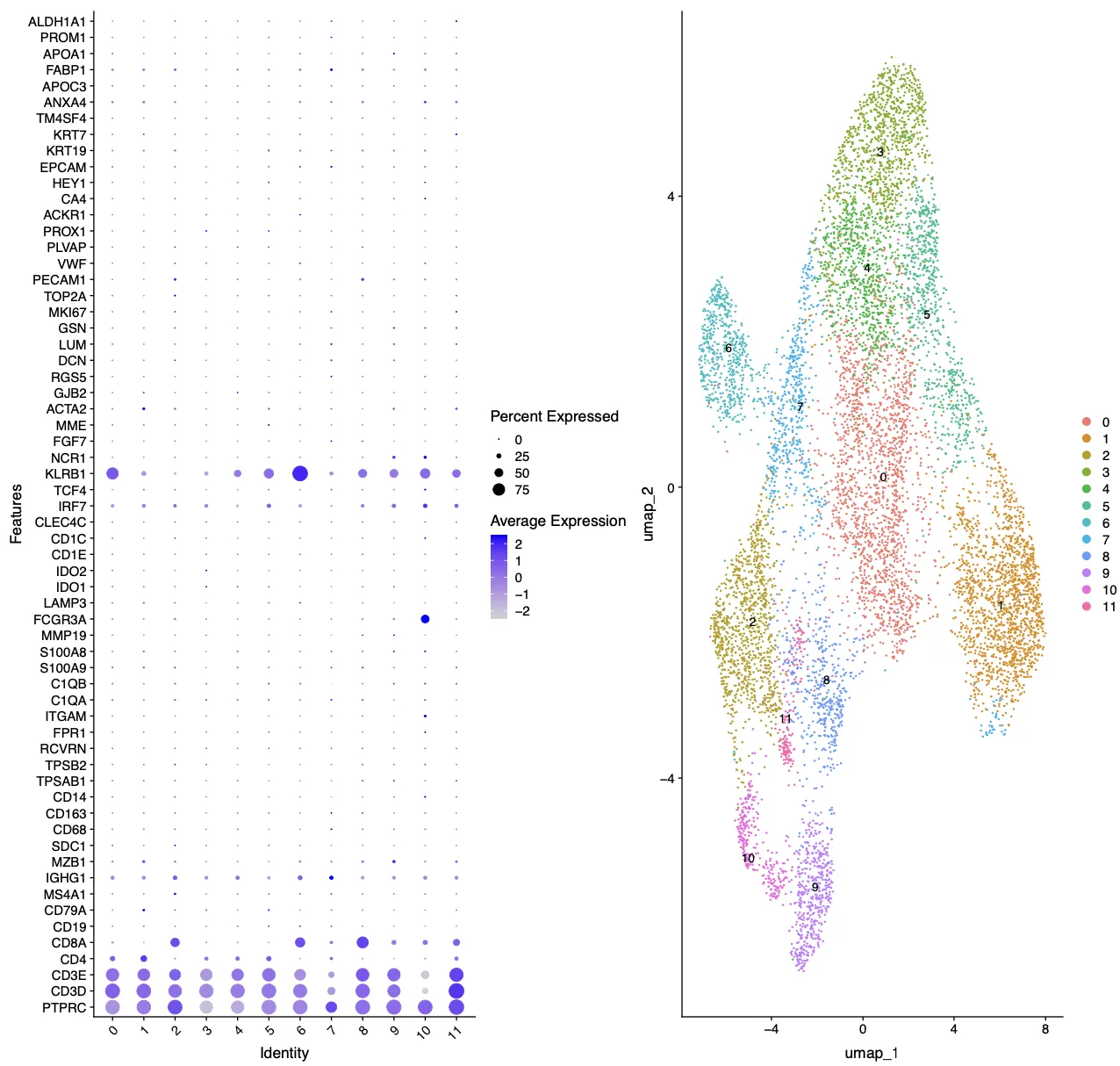
接着我们首先查看last_markers_and_umap结果,先是分辨率为0.6的结果,从这个结果来看,第6簇更像是B细胞,第11簇就更加奇怪了,里面包含了PECAM1, KLRB1, TCF4, IRF7这些基因。
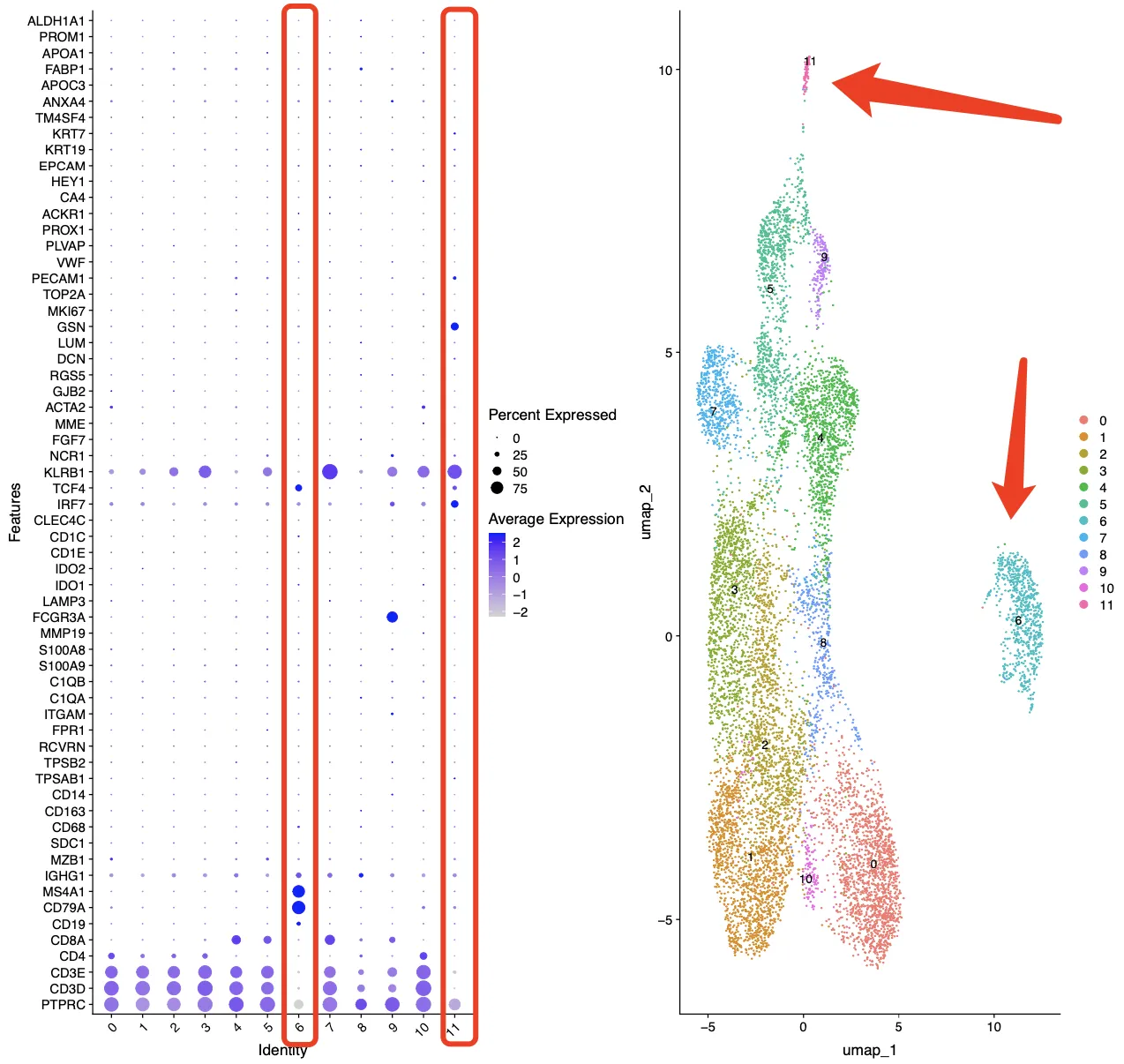
再看一下cosg分析后每个簇中的Top3基因,第6簇基本上是B细胞没跑了。

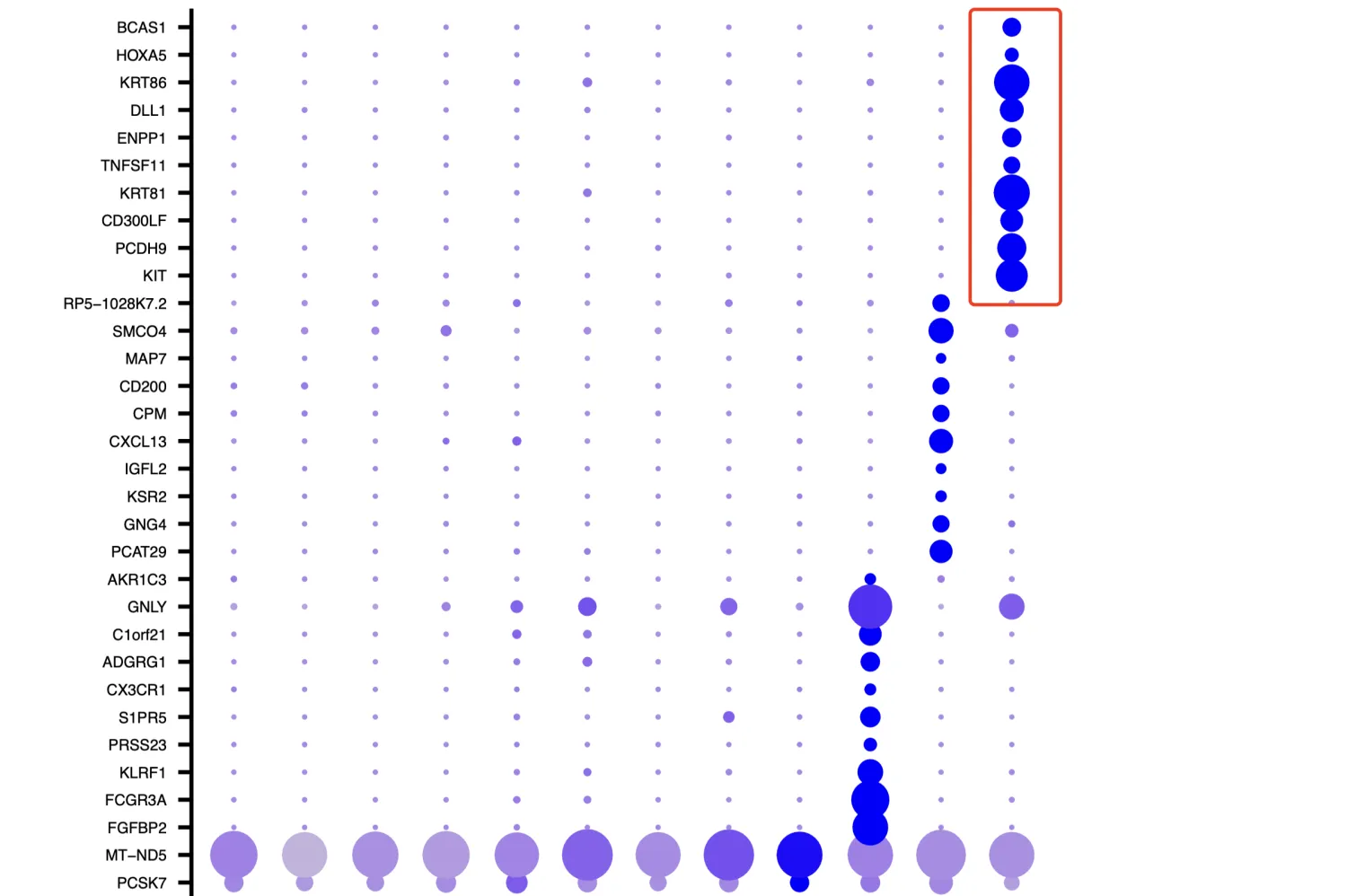
再看一下cosg分析后每个簇中的Top10基因,第11簇细胞高表达BCAS1, HOXA5, KRT86, DLL1, ENPP1, TNFSF11, KRT81,CD300LF, PCDH9, KIT,这里面也不存在免疫细胞相关的标志物。再综合上面的UMAP图,11簇的细胞含量应该是非常少的,并且位置虽然与核心大群靠的比第6簇近,但也确实没有混合在一起。因此综合上面的信息考虑,第11簇很可能是一群“混杂”细胞,具体是什么笔者也不知道。
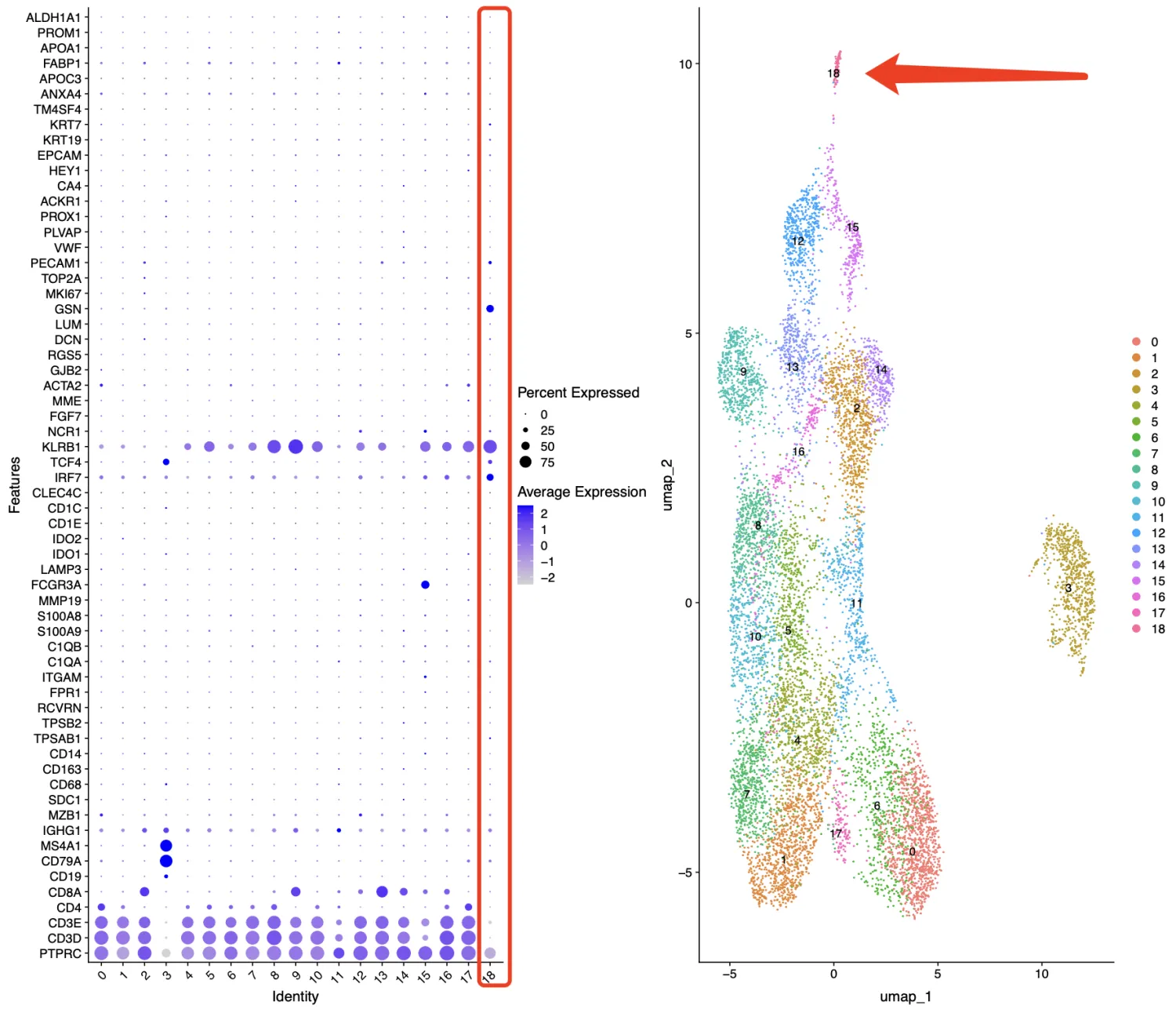
当然我们还需要考虑一件事,是不是因为我们的分辨率不够,导致第11簇没有分开,因此笔者特异会多查看几个标记基因库。比如上面设定了分辨率为1.5后我们再查看last_markers_and_umap结果,奇怪的marker组合还是存在的,因此跟分辨率无关。
3.第二步:粗分T/NK细胞并进行细胞"清洗"
3.1 清洗细胞T/NK细胞
这里是衔接上一步的内容
Idents(sce) <- sce$RNA_snn_res.0.6
table(sce@active.ident)
sub_data <- sce[,!Idents(sce) %in% c("6","11")]
table(sub_data@active.ident)
# check
DimPlot(sub_data,reduction = "umap",
label = TRUE, pt.size = 0.5)
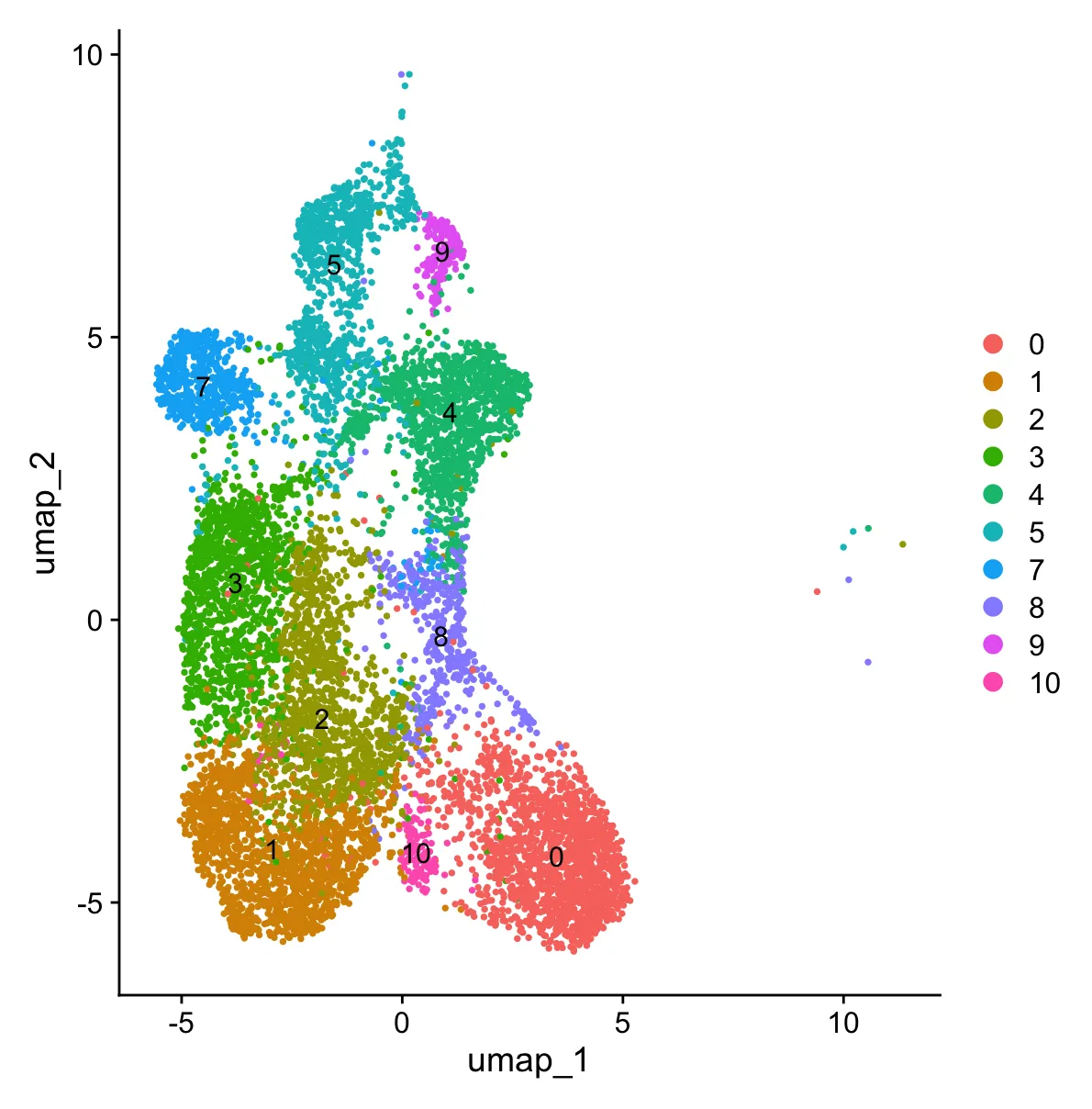
3.2 再次标准流程
标准流程就跟上面的一模一样(也可用管道符写成一小段),具体代码不再重复展示。现在已经进入细分阶段了,因此在选择分辨率时,笔者倾向于中高分辨率(下图是跑流程后的结果)。
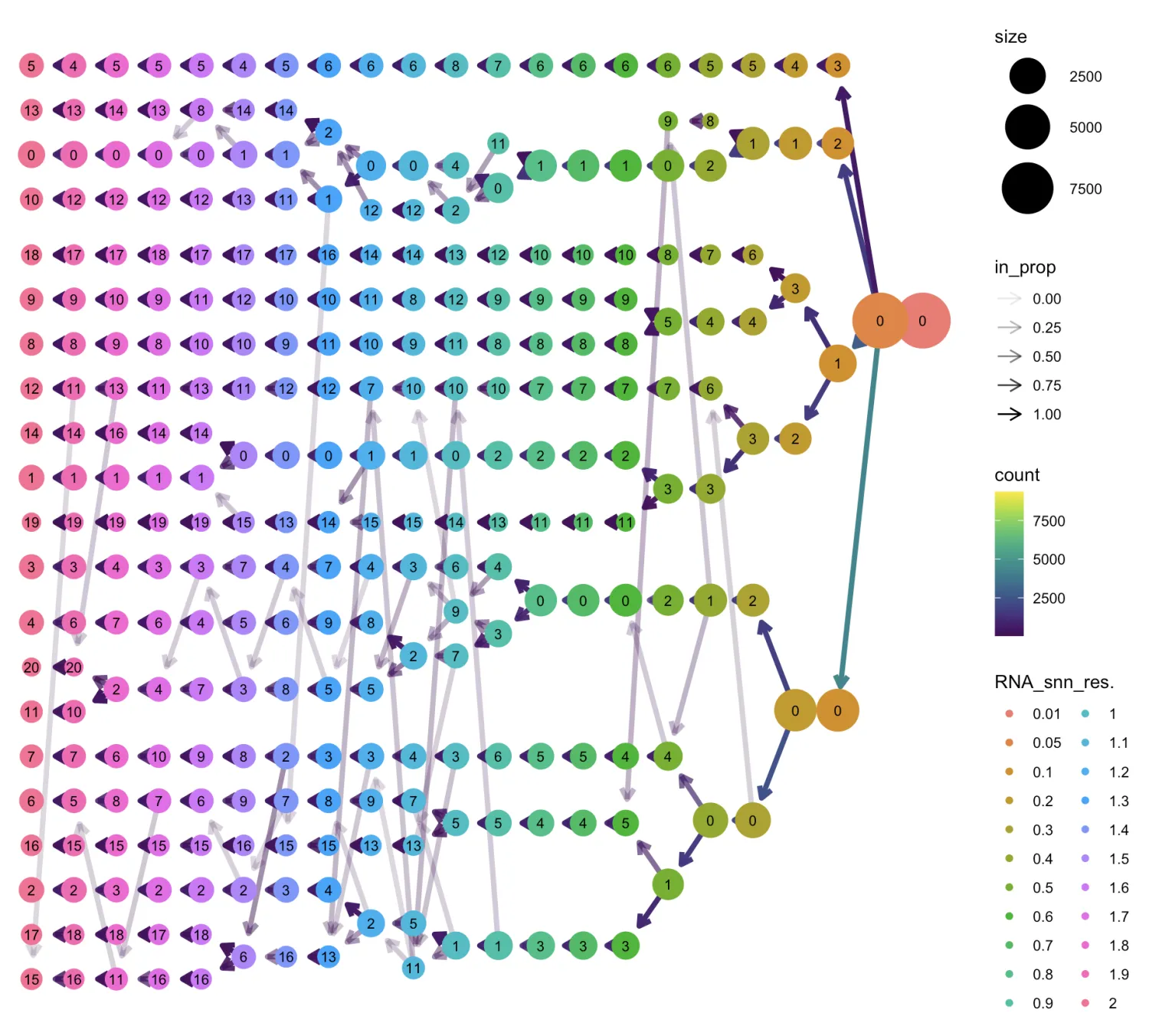
3.3 查看标志物
sce.all.int <- sce
sp = "human"
dir.create('check-by-0.6-1')
setwd('check-by-0.6-1')
sel.clust = "RNA_snn_res.0.6"
sce.all.int <- SetIdent(sce.all.int, value = sel.clust)
table(sce.all.int@active.ident)
source('../../scRNA_scripts/check-all-markers.R')
setwd('../')
getwd()
dir.create('check-by-1.5-1')
setwd('check-by-1.5-1')
sel.clust = "RNA_snn_res.1.5"
sce.all.int <- SetIdent(sce.all.int, value = sel.clust)
table(sce.all.int@active.ident)
source('../../scRNA_scripts/check-all-markers.R')
setwd('../')
getwd()
查看一下分辨率为1.5时的结果。
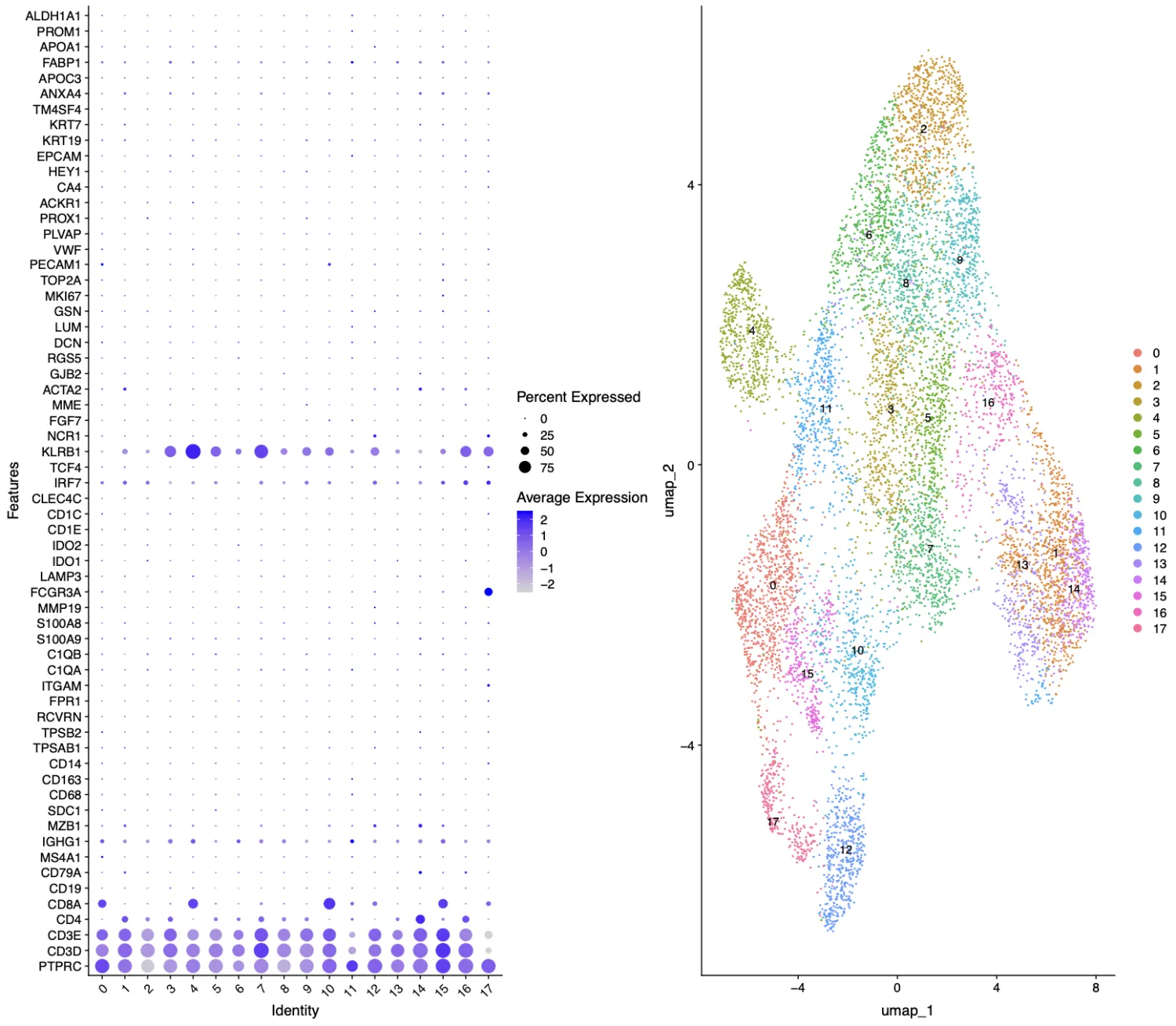
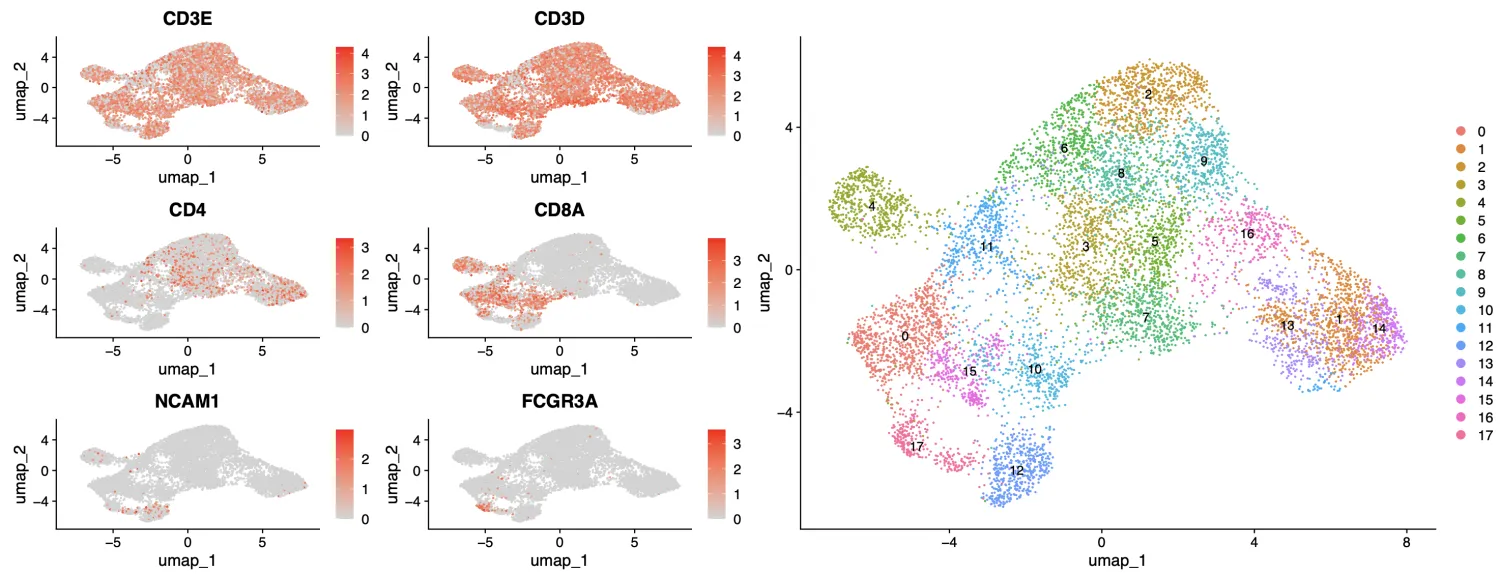
此外对于细分亚群,我们需要看一下重要标记的表达量分布,笔者根据上面clustree图的结果选择了1.5和0.6。
FeaturePlot
sel.clust = "RNA_snn_res.1.5"
sce <- SetIdent(sce, value = sel.clust)
# featurePlot可视化
marker <- c("CD3E","CD3D","CD4","CD8A","NCAM1","FCGR3A")
p <- FeaturePlot(sce,features = marker,cols = c("lightgrey" ,"red"),
combine = TRUE,raster=FALSE)
p|DimPlot(sce,label = T)
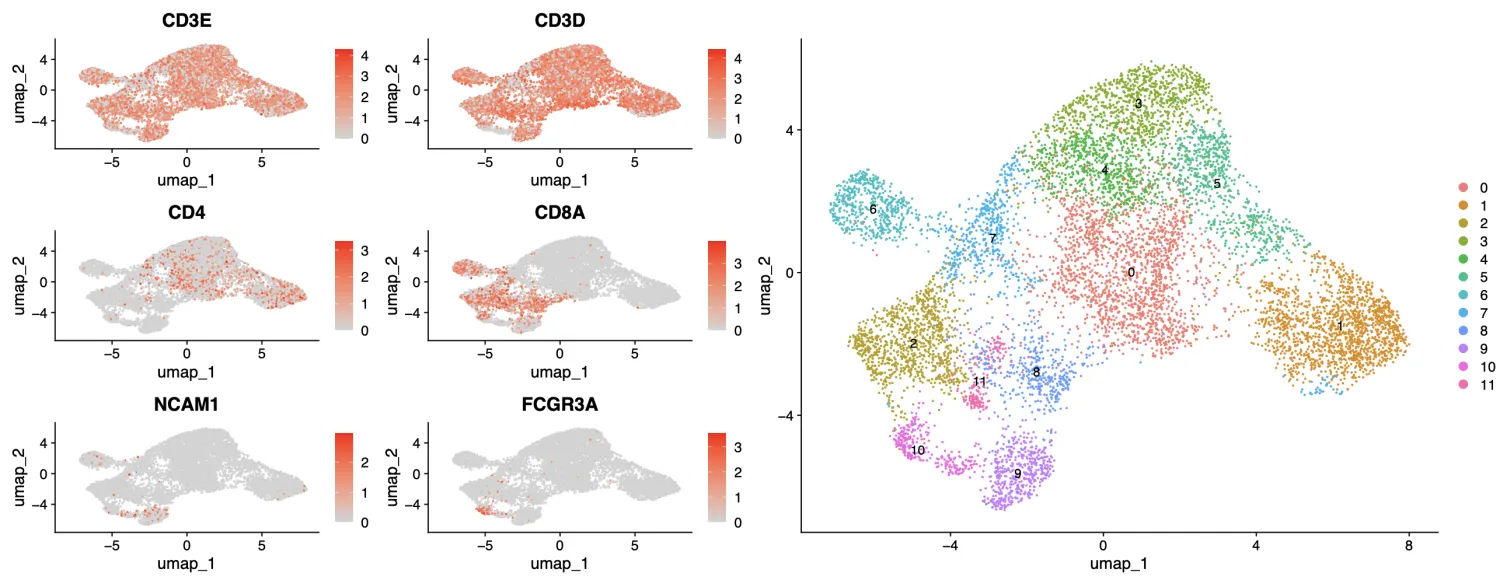
粗看会发现CD3细胞出现在每一个簇中,CD4主要分布在右上部分的簇,CD8主要在左下部分的簇,NK细胞在最左下边。
VlnPlot
# VlnPlot可视化
library(paletteer)
VlnPlot(sce,features = marker,cols = c(paletteer_d("awtools::bpalette"),
paletteer_d("awtools::a_palette"),
paletteer_d("awtools::mpalette"),
paletteer_d("awtools::spalette")),
combine = TRUE,raster=FALSE)
CD4在第1, 3, 7, 14, 16簇中比较明显聚集,CD8A在第0, 4, 10, 15簇中比较明显聚集,FCGR3A在第17簇中明显聚集。第2, 5, 6, 8, 9, 11, 12, 13簇暂时不知道是什么亚群。
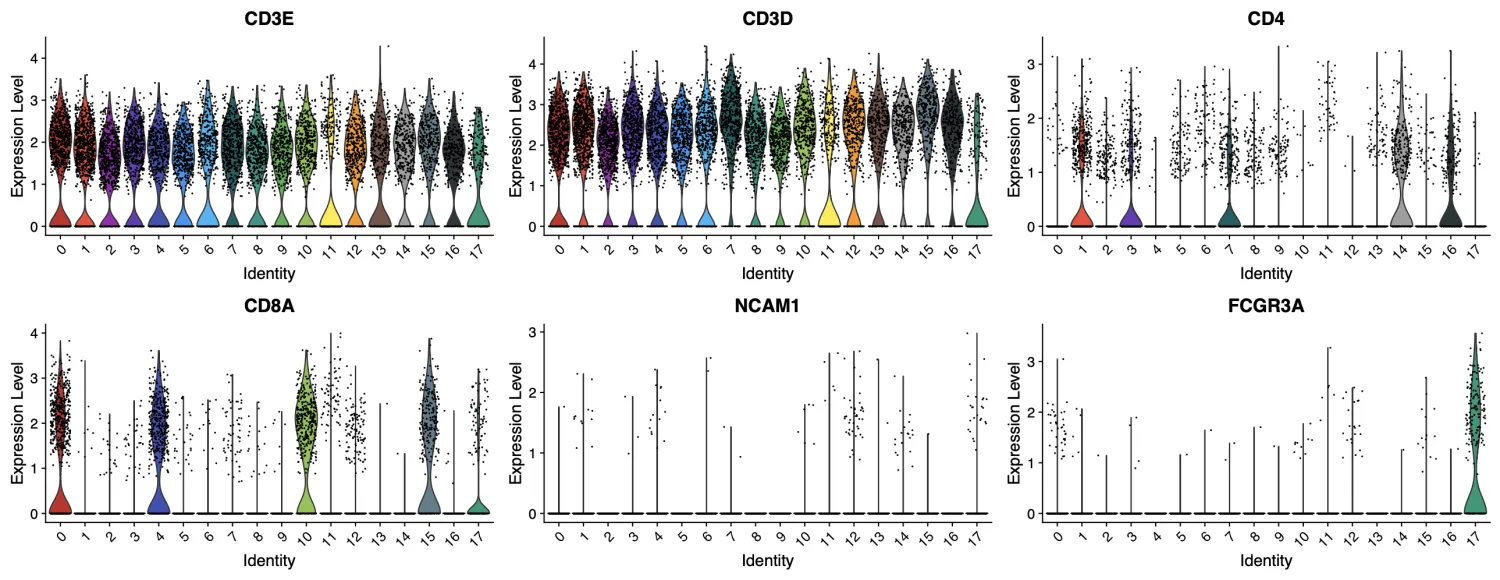
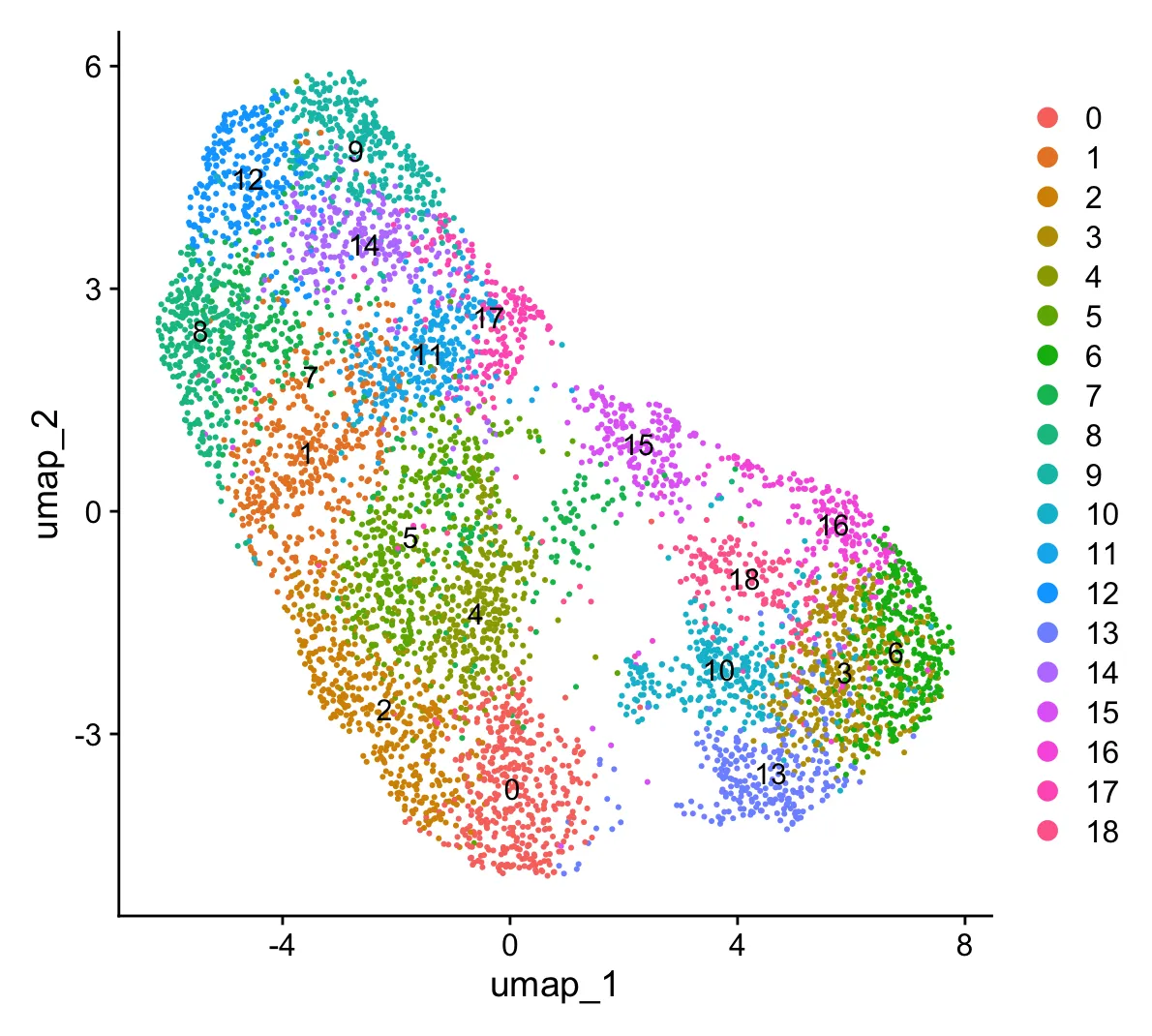
同时再看一下分辨率为0.6时的结果,我们发现第0, 1, 5簇属于CD4+T细胞,第2, 6, 8, 11簇属于CD8+T细胞,第10簇属于NK细胞,第3, 4, 7, 9簇暂时还不能够确定。
虽然本文的重点是CD4+T细胞,但由于CD4⁺T细胞的细分工作十分复杂且具有挑战性,需要在下一章中专门展开讨论,因此笔者先以NK细胞为例进行初步探讨,作为一次“小练手”,同时也检查一下NK细胞簇中是否混杂了其他的细胞。
NK细胞通常不表达CD3/CD4,理论上我们需要再对第10簇进行细分,这里分辨率选择1.5和0.6都可以,笔者选择了0.6。
同时需要做一些准备工作:要了解NK细胞发育过程中的谱系标志物变化以及分群相关的知识,首选学习教科书+文献+高分论著。后续的CD4+T细胞分群之前也需要了解其相关的知识。
sce1 <- FindSubCluster(
sce,
cluster="10",
graph.name = "RNA_snn",
subcluster.name = "RNA_snn_res.0.6_c1_sub",
resolution = 0.6
)
sel.clust = "RNA_snn_res.0.6_c1_sub"
sce1 <- SetIdent(sce1, value = sel.clust)
DimPlot(sce1,label = T)
# VlnPlot可视化
library(paletteer)
VlnPlot(sce1,features = marker,cols = c(paletteer_d("awtools::bpalette"),
paletteer_d("awtools::a_palette"),
paletteer_d("awtools::mpalette"),
paletteer_d("awtools::spalette")),
combine = TRUE,raster=FALSE)
以笔者的浅见来看,第10_2簇是classical NK细胞,第10_0簇拟命名为CD8+ NK,但我们这里不重点探查NK细胞,因此这两群细胞统一命名为NK。第10_1簇暂时命名为CD3+T细胞。
-
A CD8+ NK cell transcriptomic signature associated with clinical outcome in relapsing remitting multiple sclerosis. Nat Commun. 2021 Jan 27;12(1):635. PMID: 33504809
-
Natural Killer Cells in Cancer and Cancer Immunotherapy. Cancer Lett. 2021 Nov 1:520:233-242. PMID: 34302920
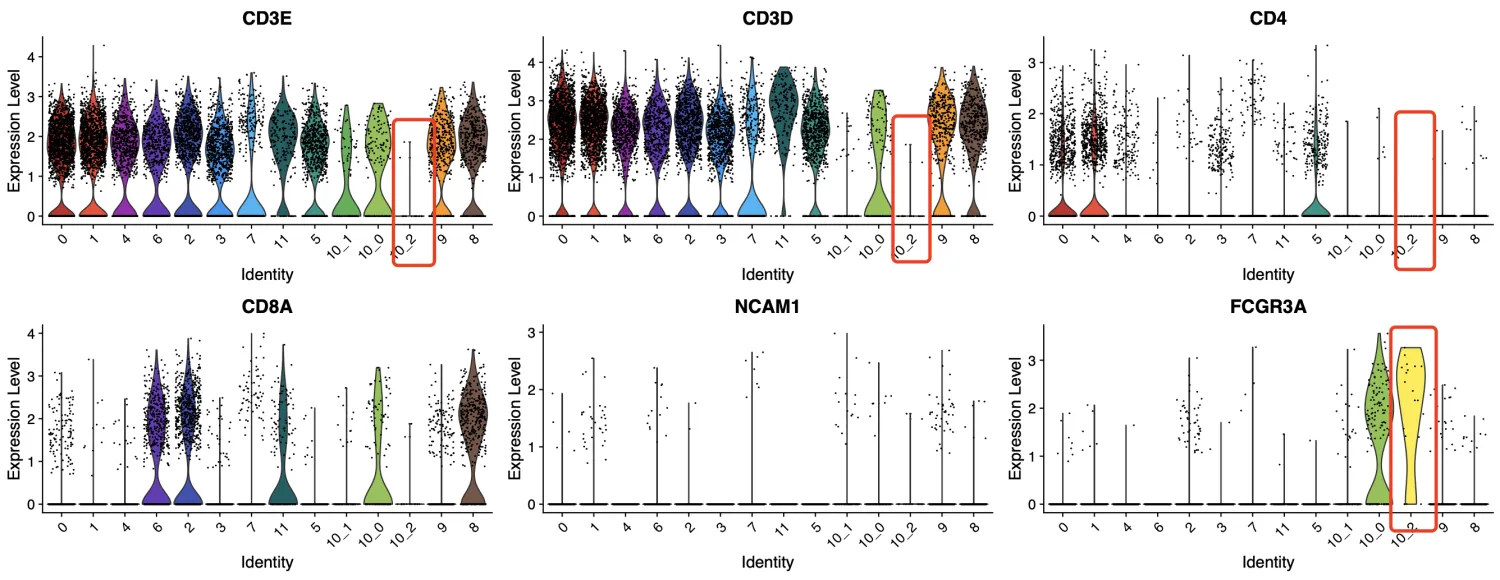
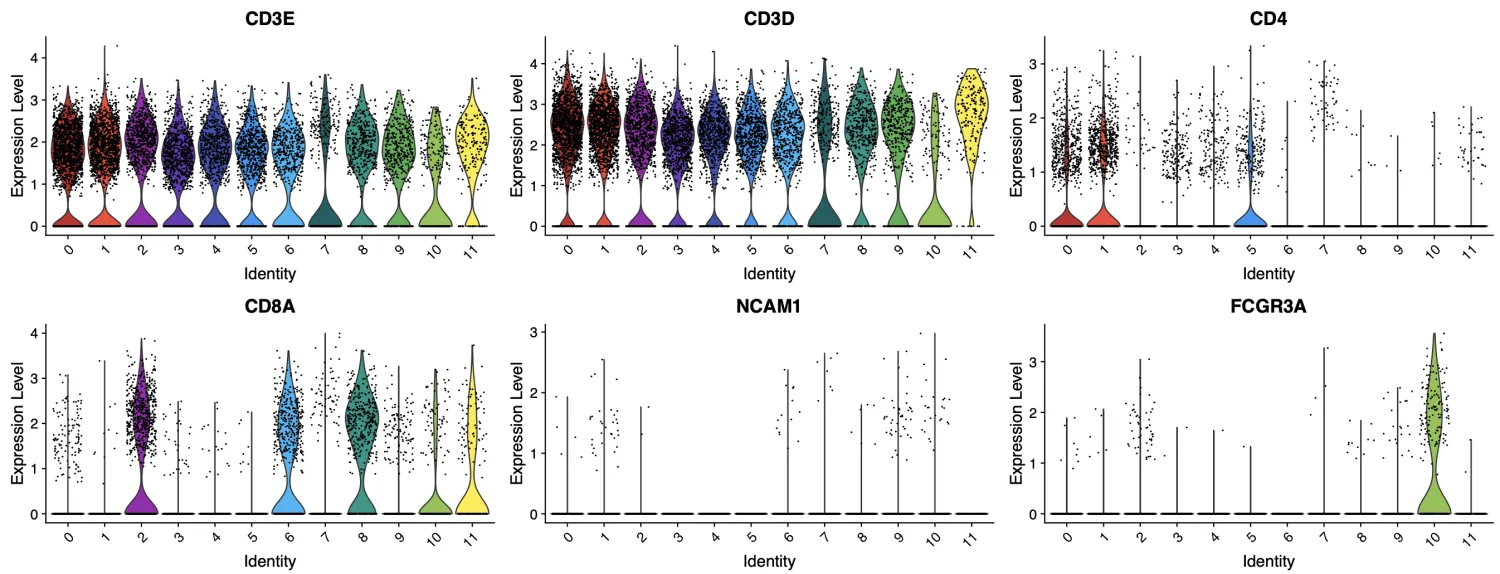
接下来,我们重点关注CD4⁺T细胞。首先来看尚未确定的第3、4、7、9簇细胞。在上述分析结果中,第3簇和第4簇的CD4表达水平均高于CD8A,且细胞数量也更多;第7簇中,CD4和CD8A的表达水平以及细胞数量都较为接近;而第9簇中,CD8A的表达水平高于CD4,且细胞数量更多。因此,我们可以初步判断:第3簇和第4簇倾向于CD4⁺T细胞,第9簇倾向于CD8⁺T细胞,而第7簇的分类暂时还不能确定。
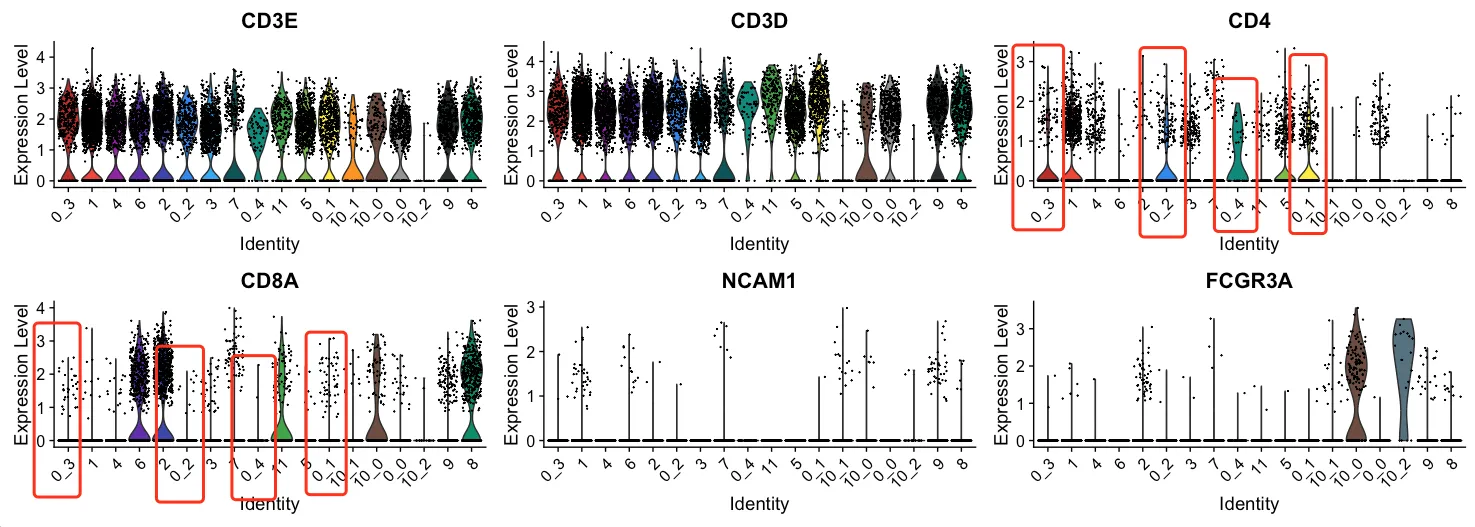
为了进一步验证这些判断并确保细胞分类的准确性,我们将使用FindSubCluster方法对细胞进行更细致的分类。此次需要重新分簇的细胞簇编号除了有3、4、7外还有0、1和5。
那么为什么还需要查看每一个细胞簇?笔者认为要确认里面是否存在一些混杂细胞,比如这里是否有可以单独分出来的CD8+T
查看第0簇
sce2 <- FindSubCluster(
sce1,
cluster="0",
graph.name = "RNA_snn",
subcluster.name = "RNA_snn_res.0.6_c1_sub",
resolution = 0.6
)
sel.clust = "RNA_snn_res.0.6_c1_sub"
sce2 <- SetIdent(sce2, value = sel.clust)
DimPlot(sce2,label = T)
# VlnPlot可视化
library(paletteer)
marker <- c("CD3E","CD3D","CD4","CD8A","NCAM1","FCGR3A")
VlnPlot(sce2,features = marker,cols = c(paletteer_d("awtools::bpalette"),
paletteer_d("awtools::a_palette"),
paletteer_d("awtools::mpalette"),
paletteer_d("awtools::spalette")),
combine = TRUE,raster=FALSE)
经过精细分群后,我们发现每个亚群中主要表达CD4,仅少量表达CD8A。从这个结果来看,第1簇无需进行进一步的精细分群,因为其中没有需要清洗的非CD4⁺细胞。
或许有人会质疑:既然这些细胞中还表达了一定量的CD8A,是否还能将其归类为CD4⁺T细胞?答案是肯定的。生物学是一个动态过程,T细胞的生长、发育和分化过程中,标记物的表达是逐渐变化的,不可能完全“泾渭分明”。
基于同样的逻辑,我们对第1、3、4、5簇也进行了类似的探查,结果与第0簇相似,均显示主要表达CD4,仅少量表达CD8A。
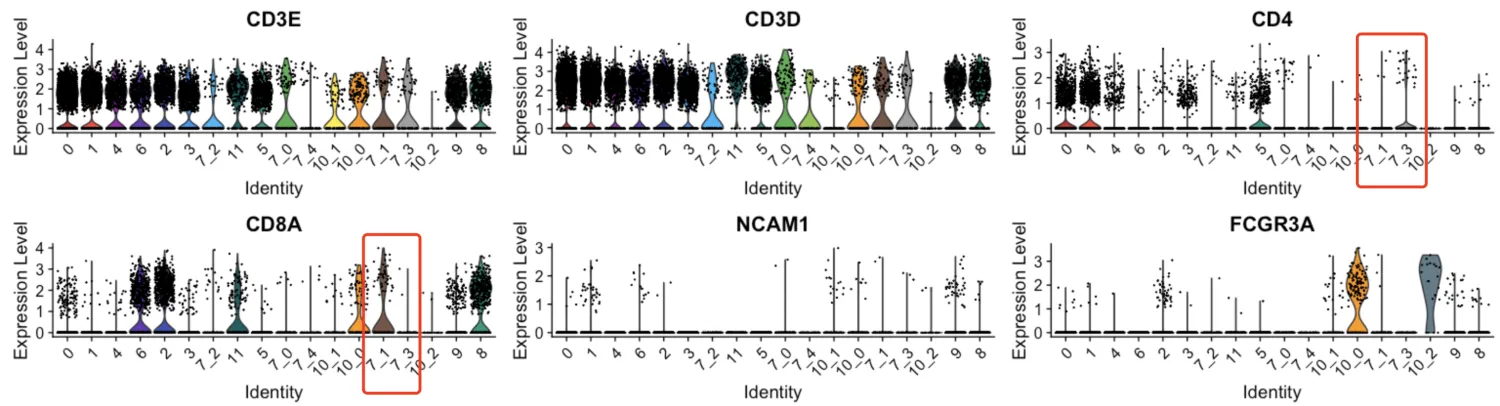
查看第7簇
sce2 <- FindSubCluster(
sce1,
cluster="7",
graph.name = "RNA_snn",
subcluster.name = "RNA_snn_res.0.6_c1_sub",
resolution = 0.6
)
sel.clust = "RNA_snn_res.0.6_c1_sub"
sce2 <- SetIdent(sce2, value = sel.clust)
DimPlot(sce2,label = T)
# VlnPlot可视化
library(paletteer)
marker <- c("CD3E","CD3D","CD4","CD8A","NCAM1","FCGR3A")
VlnPlot(sce2,features = marker,cols = c(paletteer_d("awtools::bpalette"),
paletteer_d("awtools::a_palette"),
paletteer_d("awtools::mpalette"),
paletteer_d("awtools::spalette")),
combine = TRUE,raster=FALSE)
第7_1簇可分到CD8+T细胞中去,第7_3簇分到CD4+T细胞中去,第7_0, 7_2,7_4暂定为CD3+T细胞
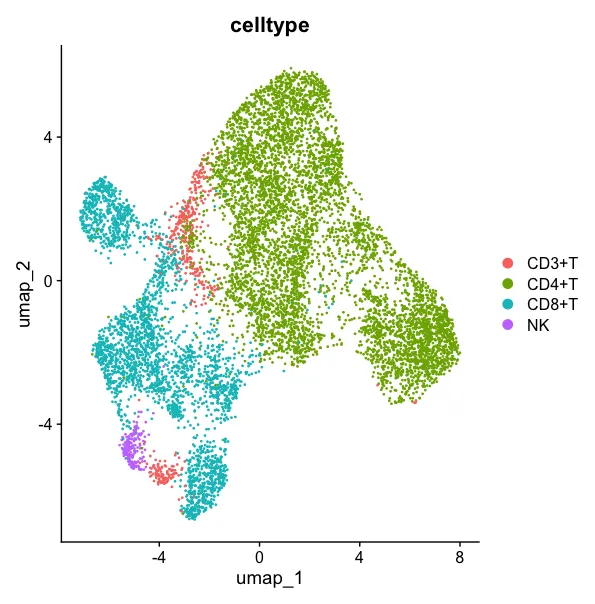
3.4 细胞命名
table(sce2$RNA_snn_res.0.6_c1_sub)
#####细胞生物学命名
celltype <- data.frame(
ClusterID = c(0:6, "7_0", "7_1", "7_2", "7_3", "7_4", 8, 9, "10_0", "10_1", "10_2", 11),
celltype = c(0:6, "7_0", "7_1", "7_2", "7_3", "7_4", 8, 9, "10_0", "10_1", "10_2", 11) # 填充 celltype 列的值
)
# 这里强烈依赖于生物学背景
celltype[celltype$ClusterID %in% c(2,6,8,9,11,"7_1" ),2]='CD8+T'
celltype[celltype$ClusterID %in% c(0,1,5,3,4,"7_3" ),2]='CD4+T'
celltype[celltype$ClusterID %in% c("10_2","10_0" ),2]='NK'
celltype[celltype$ClusterID %in% c("10_1","7_0","7_2","7_4" ),2]='CD3+T'
table(celltype$celltype)
table(sce2@meta.data$RNA_snn_res.0.6_c1_sub)
table(celltype$celltype)
sce2@meta.data$celltype = "NA"
# 记得修改RNA_snn_res.0.6
for(i in 1:nrow(celltype)){
sce2@meta.data[which(sce2@meta.data$RNA_snn_res.0.6_c1_sub == celltype$ClusterID[i]),'celltype'] <- celltype$celltype[i]}
table(sce2@meta.data$celltype)
DimPlot(sce2,group.by = "celltype")
这个环节还需要反复探查一下细胞数的问题,虽然细胞数不同文章中的异质性很大,但分析人员自己要做到心中有数。
3.5.最后提取CD4+T细胞亚群,并重新走一遍标准化流程
标准化流程如上或者自己准备一个简单的即可
Idents(sce2) <- sce2$celltype
table(sce2@active.ident)
sub_data <- sce2[,Idents(sce2) %in% c("CD4+T")]
table(sub_data@active.ident)
# check
DimPlot(sub_data,reduction = "umap",
label = TRUE, pt.size = 0.5)
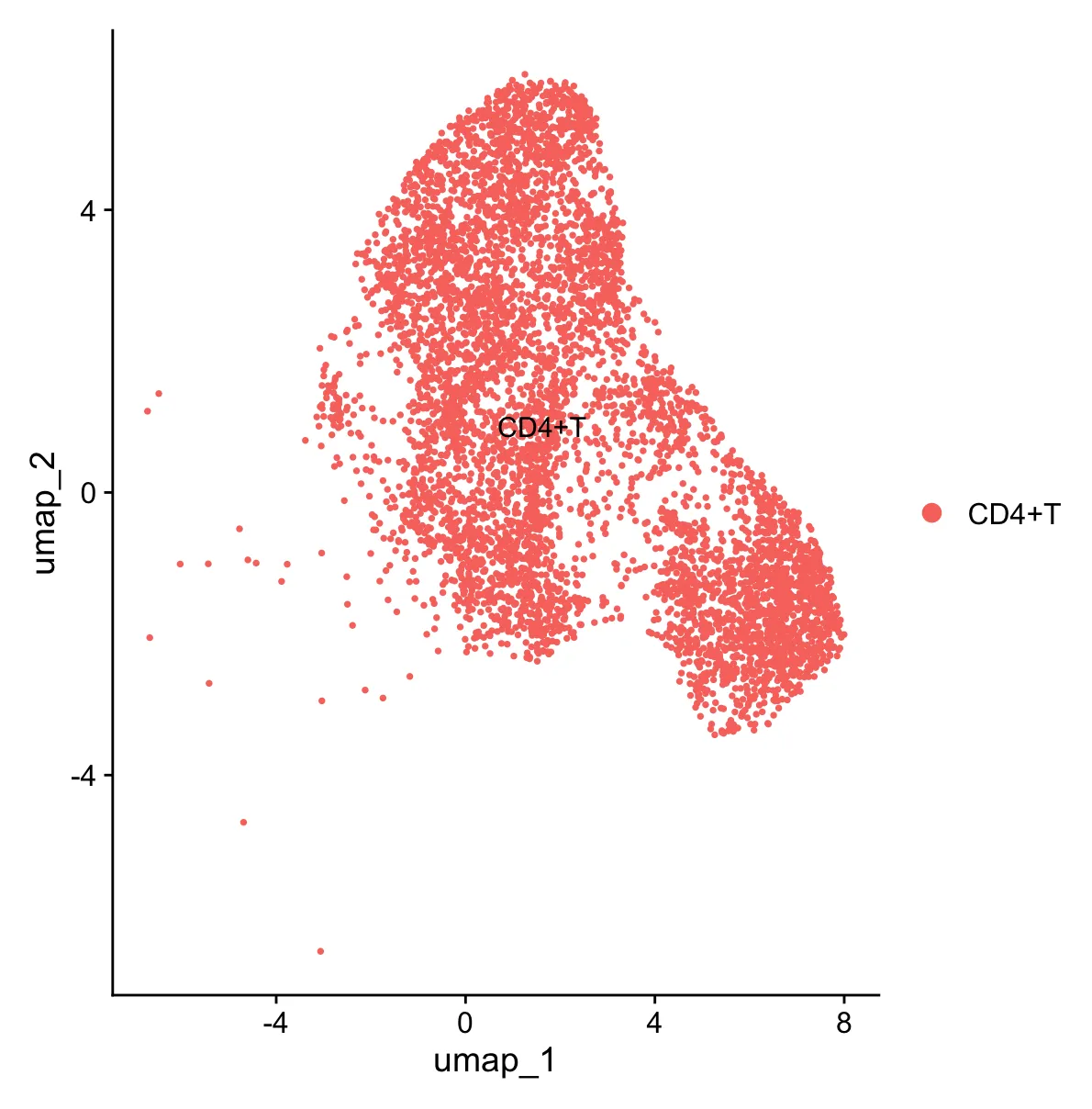
走完标准化流程之后的UMAP分群图
3.6.保存数据
qsave(sce,"sub_data_cd4+T.qs")
setwd("..")
本次内容完成了亚细胞分群从T/NK细胞至CD4+T细胞流程的实践流程,核心思想就是如何合理地进行细胞“清洗”,同时笔者也对内容做一定的总结。
-
亚群细分也应遵循逐级细分流程,比如先从T/NK细胞到CD4+T细胞,而非直接围绕T/NK进行全面的精细分群。
-
细分流程在遵照初级篇1中的核心内容之外,还需要对每一簇细胞单独探查并清洗。
-
在分簇过程中,要用多种表达量展示工具去展示关键markers在不同簇中分布和表达情况。
-
一定要有生物学相关知识,建议教科书+综述+高分论著。
参考资料:
-
SIB-Swiss Institute of Bioinformatics: https://www.youtube.com/watch?v=kqcvqqZp3Jo
-
Comprehensive Phenotyping of T Cells Using Flow Cytometry. Cytometry A. 2019 Jun;95(6):647-654. PMID: 30714682
-
A CD8+ NK cell transcriptomic signature associated with clinical outcome in relapsing remitting multiple sclerosis. Nat Commun. 2021 Jan 27;12(1):635. PMID: 33504809
-
Natural Killer Cells in Cancer and Cancer Immunotherapy. Cancer Lett. 2021 Nov 1:520:233-242. PMID: 34302920
致谢:感谢曾老师以及生信技能树团队全体成员。更多精彩内容可关注公众号:生信技能树,单细胞天地,生信菜鸟团等公众号。
注:若对内容有疑惑或者有发现明确错误的朋友,请联系后台(欢迎交流)。更多相关内容可关注公众号:生信方舟
- END -

















 1万+
1万+

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









