







目前已完成基于R语言的单细胞分析实战系列,涵盖初级、中级和高级三个阶段(每个阶段暂时各包含4篇内容,后续将陆续更新)。接下来,将尝试开展基于Python的单细胞分析实战系列。但在系列内容正式开始之前,笔者认为有必要先学习一下数据转化和读取流程。
本次内容涉及到的工程文件可通过网盘获得:中级篇2,链接: https://pan.baidu.com/s/1y-HHLXoXsJbgWKCdz26-gQ 提取码: yx93 。
此外,可以向“生信技能树”公众号发送关键词‘单细胞’,直接获取Seurat V5版本的完整代码。
首先推荐python官网:https://www.python.org/doc/
里面的教程有多语言版本,可以选择中文
快速检索就可以查询想要的信息,比如查了一下os库
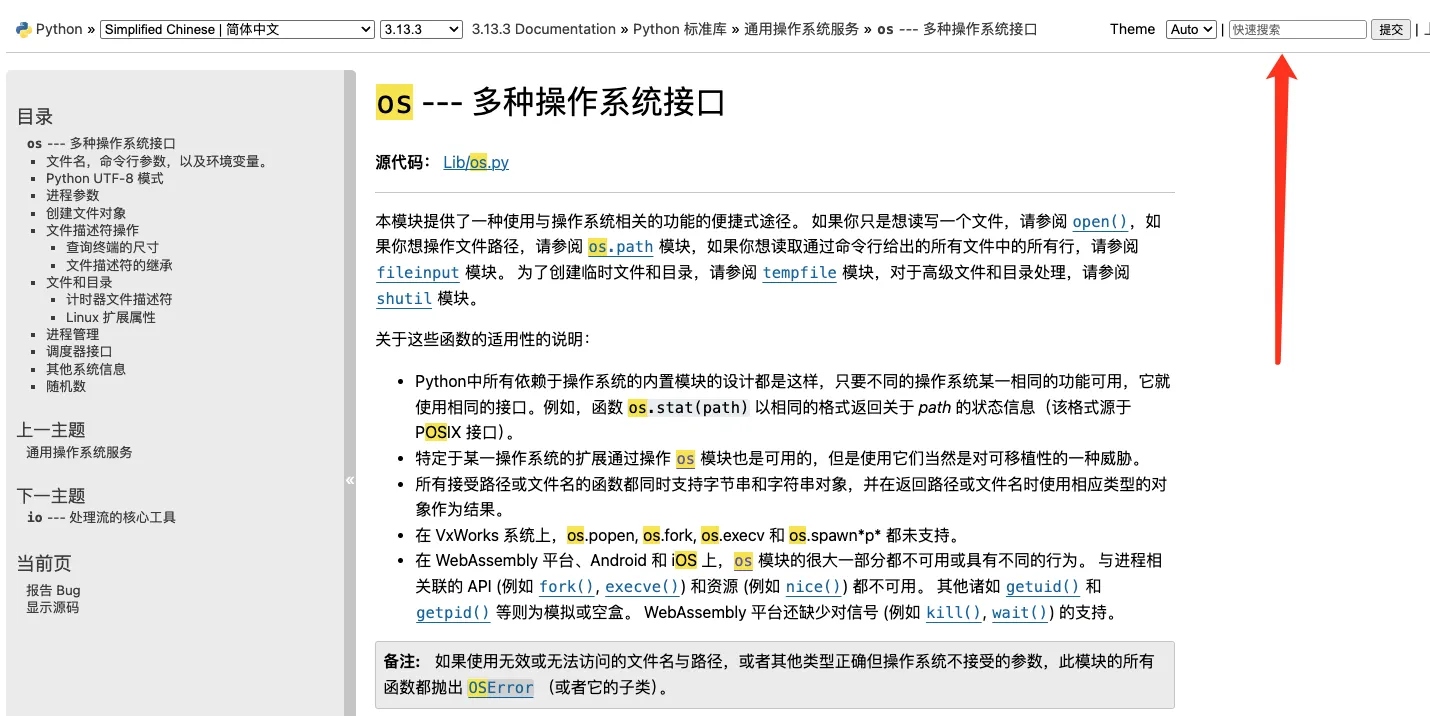
这个网站可以作为工具手册帮助使用者快速上手,当然大模型辅助也一定是必不可少的。
Seurat和h5ad数据相互转化
后续一定会涉及到数据的相互转化,因此必须学会这个步骤。
1.导入(Seurat V5对象转换为h5ad数据)
R语言部分
rm(list = ls())
library(Seurat)
library(qs)
library(reticulate)
library(hdf5r)
library(sceasy)
library(BiocParallel)
register(MulticoreParam(workers = 4, progressbar = TRUE))
scRNA_V5 <- qread("CD4+T_final.qs")
scRNA_V5
# An object of class Seurat
# 21308 features across 5912 samples within 1 assay
# Active assay: RNA (21308 features, 2000 variable features)
# 3 layers present: counts, data, scale.data
# 3 dimensional reductions calculated: pca, harmony, umap
2.配置python环境
linux/终端,numpy不能使用最新版本
conda create -n sceasy python=3.9
conda activate sceasy
conda install anndata
conda install scipy
conda install loompy
conda install numpy==1.24.4
3.R语言转换
再次回到R语言, 在R语言中调用Python环境
# 在R语言中加载python环境
use_condaenv('sceasy')
loompy <- reticulate::import('loompy')
py_config()
# python: /opt/anaconda3/envs/sceasy/bin/python
# libpython: /opt/anaconda3/envs/sceasy/lib/libpython3.9.dylib
# pythonhome: /opt/anaconda3/envs/sceasy:/opt/anaconda3/envs/sceasy
# version: 3.9.20 | packaged by conda-forge | (main, Sep 22 2024, 14:06:09) [Clang 17.0.6 ]
# numpy: /opt/anaconda3/envs/sceasy/lib/python3.9/site-packages/numpy
# numpy_version: 1.24.4
# loompy: /opt/anaconda3/envs/sceasy/lib/python3.9/site-packages/loompy
开始转换
# Seurat to AnnData
scRNA_V5[["RNA"]] <- as(scRNA_V5[["RNA"]], "Assay")
sceasy::convertFormat(scRNA_V5, from="seurat", to="anndata",
outFile='scRNA_V5.h5ad')
# AnnData object with n_obs × n_vars = 5042 × 20124
# obs: 'nCount_RNA', 'nFeature_RNA', 'Sample', 'Cell.Barcode', 'Type', 'RNA_snn_res.0.1', 'RNA_snn_res.0.2', 'RNA_snn_res.0.3', 'RNA_snn_res.0.4', 'RNA_snn_res.0.5', 'RNA_snn_res.0.6', 'RNA_snn_res.0.7', 'RNA_snn_res.0.8', 'RNA_snn_res.0.9', 'RNA_snn_res.1', 'RNA_snn_res.1.1', 'RNA_snn_res.1.2', 'seurat_clusters', 'celltype', 'seurat_annotation'
# var: 'vf_vst_counts_mean', 'vf_vst_counts_variance', 'vf_vst_counts_variance.expected', 'vf_vst_counts_variance.standardized', 'vf_vst_counts_variable', 'vf_vst_counts_rank', 'var.features', 'var.features.rank'
# obsm: 'X_pca', 'X_harmony', 'X_umap'
# Warning message:
# In .regularise_df(obj@meta.data, drop_single_values = drop_single_values) :
# Dropping single category variables:orig.ident
4.在jupyter lab中确认一下
打开jupyter lab
首先必须要安装anaconda/miniconda等;
-
直接点击电脑上的anaconda软件(笔者装的事anaconda所以这里这么写了),进入界面之后点击jupyter lab

-
在终端中输入jupyter lab,电脑会自动打开该界面,如果直接以家为自己的工作目录,就直接输入jupyter lab,如果想要切换工作目录,那就要先cd进去,再输入jupyter lab
# mkdir ~/xxxx
cd ~/xxxx
jupyter lab
注册python内核
激活环境之后,注册小环境对应的内核, 在Jupyter Notebook中运行Python代码时,真正执行这些代码的“引擎”就是ipykernel,Jupyter 提供的是前端界面(网页 Notebook),ipykernel是后端:它运行你的代码,并把结果返回给前端显示。所以先要安装一下ipykernel。
conda activate sceasy
pip install ipykernel
python -m ipykernel install --user --name sceasy --display-name "Python(sceasy)"
增加了一个新的sceasy内核,以后涉及到数据转化就可以连接该内核。
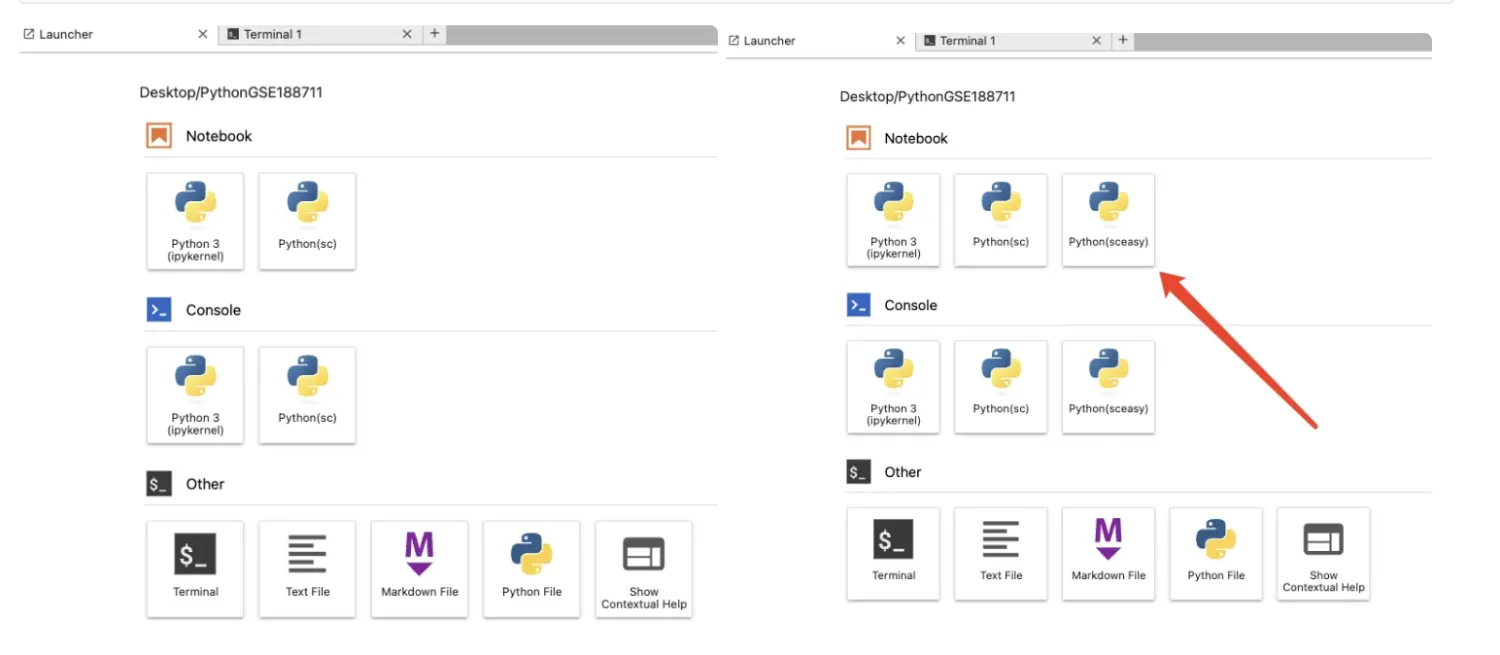
如何删除
# 查看当前列表
jupyter kernelspec list
# 删除
jupyter kernelspec uninstall sceasy
# 删除conda环境
conda remove --name sceasy --all
为什么创建了环境之后还需要注册内核?
在回答这个问题前,需要了解一下Conda、Anaconda、Miniconda
-
Conda: 一个开源的软件包管理和环境管理系统,最初是为了支持Python语言而设计的,但现在也可以管理和部署任何语言的软件包和依赖项。它能够帮助用户轻松地安装不同的软件版本,并且可以在相互隔离的环境中管理这些软件包,从而避免版本冲突。Conda 可以通过命令行使用,并且支持多个平台(Windows、macOS 和 Linux)。
-
Anaconda: 基于Conda的一个发行版,专为数据科学和机器学习等任务设计。它包括了Conda以及大量的预装软件包、库和工具,比如Jupyter Notebook、Spyder IDE 等。Anaconda发行版非常适合刚开始从事数据分析、科学计算、机器学习等领域的新手,因为它几乎包含了所有常用的库和工具,减少了单独安装配置的时间。然而,由于其庞大的体积(通常超过数GB),对于只需要少量特定工具或库的用户来说,可能会显得过于臃肿。
-
Miniconda: 是一个轻量级的Conda安装器,只包含Conda包管理器和Python基础环境。相比完整的 Anaconda 发行版,Miniconda更加精简,允许用户根据自己的需求自定义安装所需的软件包和库。这种方式对于那些已经有特定要求或者希望从头开始构建自己环境的高级用户来说非常合适。Miniconda 提供了一个灵活的基础,用户可以在此基础上通过 Conda 来安装任何需要的包,而不必携带不必要的预装软件。 所以Anaconda和miniconda都是属于conda,并且conda更像是一个管理平台(工具),管理着软件和环境。
而Conda环境是一个独立的Python环境,包含特定版本的Python和安装的库,旨在隔离项目依赖和轻松管理依赖。不同项目可能需要不同版本的Python或第三方库,通过创建独立的Conda环境可以避免这些依赖之间的冲突,并允许为每个环境单独安装、更新或删除库而不影响其他环境。Jupyter Notebook或Jupyter Lab是一个交互式编程工具,它本身并不知道Conda环境的存在。为了让Jupyter能够使用某个 Conda环境中的Python和库,需要将该环境注册为一个Jupyter内核。内核是Jupyter的执行引擎,每个 Jupyter实例都会绑定到一个内核,负责运行代码并返回结果,个人理解这里的内核相当于特定扇门或者钥匙把jupyter lab和conda环境打通。即使已经创建了一个 Conda环境,Jupyter并不会自动识别这个环境。因此,需要手动将Conda环境注册为Jupyter内核,以便在Jupyter中选择特定环境对应的内核,确保代码运行在正确的环境中,并能够访问该环境中安装的库(如 pandas、numpy 等)。**再如果觉得迷糊也没关系,简单来说相当于conda环境和jupyter内核其实是互相独立的,使用者只需要知道创建了环境之后再注册一下内核这样就能让他们"步调一致"即可(不然会调用base中的库)**。
在python中确认一下h5ad文件
安装一些必要的库
# 安装scanpy
pip install scanpy
加载库
import scanpy as sc
import os
# 确认路径
os.getcwd()
# '/Users/zaneflying/Desktop/PythonGSE188711'
读取数据
# 读取数据
adata = sc.read_h5ad('scRNA_V5.h5ad')
adata
# AnnData object with n_obs × n_vars = 5912 × 21308
# obs: 'orig.ident', 'nCount_RNA', 'nFeature_RNA', 'percent_mito', 'percent_ribo', 'percent_hb', 'seurat_clusters', 'RNA_snn_res.0.1', 'RNA_snn_res.0.2', 'RNA_snn_res.0.3', 'RNA_snn_res.0.5', 'RNA_snn_res.0.8', 'RNA_snn_res.1', 'celltype', 'location', 'pANN_0.25', 'contamination', 'RNA_snn_res.0.4', 'RNA_snn_res.0.6', 'RNA_snn_res.0.7', 'RNA_snn_res.0.9', 'RNA_snn_res.1.1', 'RNA_snn_res.1.2', 'RNA_snn_res.1.3', 'RNA_snn_res.1.4', 'RNA_snn_res.1.5', 'RNA_snn_res.1.6', 'RNA_snn_res.1.7', 'RNA_snn_res.1.8', 'RNA_snn_res.1.9', 'RNA_snn_res.2'
# var: 'vf_vst_counts_mean', 'vf_vst_counts_variance', 'vf_vst_counts_variance.expected', 'vf_vst_counts_variance.standardized', 'vf_vst_counts_variable', 'vf_vst_counts_rank', 'var.features', 'var.features.rank'
# obsm: 'X_harmony', 'X_pca', 'X_umap'
5.导入(h5ad数据转换为Seurat对象)
R语言部分
rm(list = ls())
library(sceasy)
library(qs)
library(reticulate)
library(Seurat)
library(BiocParallel)
register(MulticoreParam(workers = 4, progressbar = TRUE))
6.数据转换
# h5ad转为Seurat
sceasy::convertFormat(obj = "scRNA_V5.h5ad",
from="anndata",to="seurat",
outFile = 'scRNA_CD4.rds')
# X -> counts
# An object of class Seurat
# 21308 features across 5912 samples within 1 assay
# Active assay: RNA (21308 features, 0 variable features)
# 2 layers present: counts, data
# 3 dimensional reductions calculated: harmony, pca, umap
7.数据确认
scRNA <- readRDS("./scRNA_CD4.rds")
rownames(scRNA)
colnames(scRNA)

数据读取流程(Python)
1.创建和激活环境
# 创建一名为sc的python版本为3.9的环境
# -n是 -name的简称
conda create -n sc python=3.9
# 激活
conda activate sc
2.在Jupyter lab中创建内核
python -m ipykernel install --user --name sc --display-name "Python(sc)"
3.安装库
conda install scanpy
正式移动和读取文件(Python)
1.导入所需要的库
import os
import shutil
import re
2.查询当前的路径
os.getcwd()
#'/Users/zaneflying/Desktop/PythonGSE188711'
3.列出目录下所有包含 'features' 的文件
fs = [
os.path.join('GSE188711_RAW', f) # 拼接出完整路径,如 GSE188711_RAW/GSM5688707_features.tsv.gz
for f in os.listdir('GSE188711_RAW') # 遍历目录下的所有文件名
if 'features' in f # 只选取文件名中包含 'features' 的文件
]
print(fs)
samples1 = fs
# ['GSE188711_RAW/GSM5688707_features_JCA.tsv.gz', 'GSE188711_RAW/GSM5688706_features_WGC.tsv.gz',
# 'GSE188711_RAW/GSM5688711_features_R_CRC4.tsv.gz', 'GSE188711_RAW/GSM5688709_features_RS-CRC1.tsv.gz',
# 'GSE188711_RAW/GSM5688710_features_R_CRC3.tsv.gz', 'GSE188711_RAW/GSM5688708_features_LS-CRC3.tsv.gz']
4.用正则表达式替换字符串,得到样本名
samples2 = [re.sub(r'_features', '', f) for f in fs]
samples2 = [re.sub(r'\.tsv\.gz$', '', f) for f in samples2]
print(samples2)
其实这里的样本名可以清洗更加仔细,比如只剩下GSM号码,简洁的好处非常多,建议保持简洁。
samples2 = [re.findall(r'GSM\d+', f)[0] for f in fs]
print(samples2)
# ['GSM5688707', 'GSM5688706', 'GSM5688711', 'GSM5688709', 'GSM5688710', 'GSM5688708']
5.创建文件夹并重命名和移动文件
# 3. 创建 outputs 文件夹(如果不存在)
os.makedirs('outputs', exist_ok=True)
# 4. 遍历样本,创建子文件夹并复制文件
for x, y in zip(samples2, samples1):
output_dir = os.path.join('outputs', x)
os.makedirs(output_dir, exist_ok=True)
# 复制并重命名 features 文件
shutil.copy(y, os.path.join(output_dir, 'features.tsv.gz'))
# 对应的 matrix 文件名替换
matrix_file = y.replace('features', 'matrix').replace('.tsv', '.mtx')
shutil.copy(matrix_file, os.path.join(output_dir, 'matrix.mtx.gz'))
# 对应的 barcodes 文件名替换
barcodes_file = y.replace('features', 'barcodes')
shutil.copy(barcodes_file, os.path.join(output_dir, 'barcodes.tsv.gz'))
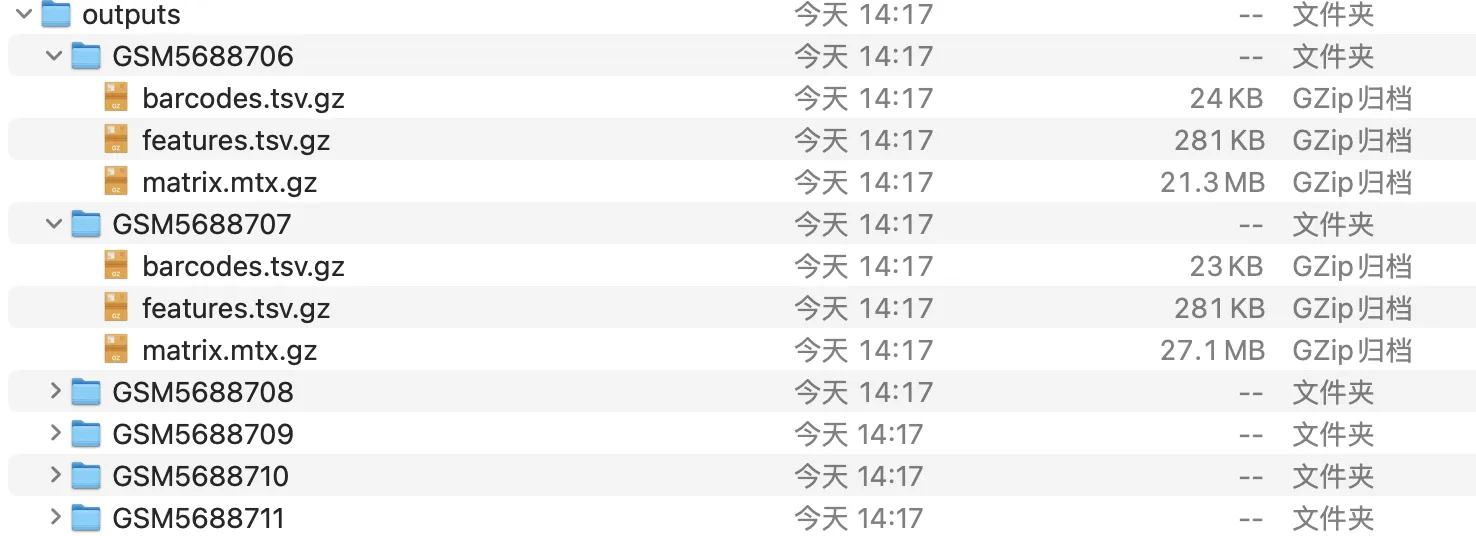
6.读取文件
6.0 安装库,linux/终端
conda install loompy
pip install numpy==1.24.4
6.1 导入
import os
import scanpy as sc
6.2 检查路径
os.getcwd()
# '/Users/zaneflying/Desktop/PythonGSE188711'
6.3 读取样本路径和信息(排除了“.”开头的隐藏文件)
dir_path = '/Users/zaneflying/Desktop/PythonGSE188711/outputs/'
samples = [s for s in os.listdir(dir_path) if not s.startswith('.')]
print(samples)
# ['GSM5688711', 'GSM5688710', 'GSM5688709', 'GSM5688707', 'GSM5688706', 'GSM5688708']
6.4 读取样本构建sce_list
sce_list = []
for sample in samples:
sample_path = os.path.join(dir_path, sample)
# 读取10X格式数据
adata = sc.read_10x_mtx(sample_path, var_names='gene_symbols', cache=True)
# 过滤:至少5个细胞的基因,至少300个基因的细胞 (对应min.cells和min.features)
sc.pp.filter_genes(adata, min_cells=5)
sc.pp.filter_cells(adata, min_genes=300)
# 给AnnData添加项目名称
adata.obs['project'] = sample
sce_list.append(adata)
# sce_list 是包含多个AnnData对象的列表
sce_list

6.5 合并
adata = sc.concat(sce_list,label='sampleid',index_unique='_')
adata.obs_names_make_unique()
adata
参数index_unique='-'帮助使用者把样本名和细胞名进行连接。
6.6 信息查看
adata.obs_names

adata.obs_keys
![]()
adata.var_names

保存h5ad文件
adata.write('GSE188711.h5ad')
保存loom文件
import loompy
adata.write_loom('GSE188711.loom')
参考资料:
-
python: https://www.python.org/doc/
-
sceasy github: https://github.com/cellgeni/sceasy
-
Anaconda: https://www.anaconda.com/
-
Jupyter: https://jupyter.org/
-
pandas: https://pandas.pydata.org/
-
os: https://docs.python.org/3/library/os.html
-
shutil:https://docs.python.org/3/library/shutil.html
-
re: https://docs.python.org/3/library/re.html
-
生信技能树:https://mp.weixin.qq.com/s/AVSRSAZDTQHHriMY2YyGOw
致谢:感谢曾老师以及生信技能树团队全体成员。更多精彩内容可关注公众号:生信技能树,单细胞天地,生信菜鸟团等公众号。
注:若对内容有疑惑或者有发现明确错误的朋友,请联系后台(欢迎交流)。更多相关内容可关注公众号:生信方舟 。

















 3279
3279

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









