








目前单细胞技术发展迅速,单细胞RNA-seq和单细胞ATAC已经应用到多个物种,植物中的多组学应用还相对较少,今天我们分享一篇单细胞ATAC+RNA-seq的文献,是由河北农业大学赵建军团队在国际知名期刊Advanced Science(中科院一区,IF:15.1)在线发表题为“Multiome in the Same Cell Reveals the Impact of Osmotic Stress on Arabidopsis Root Tip Development at Single-Cell Level”的研究论文。该研究同时检测了16670个拟南芥细胞核的基因表达和染色质可及性,重建了拟南芥根尖细胞类型特异性转录调控网络,揭示了单细胞多组方法在解析渗透胁迫响应机制中的作用。
「 研究背景 」
细胞特异性转录调控网络(TRNs)在植物发育和对环境胁迫的响应中起着至关重要的作用。通过同时检测16670个拟南芥根尖细胞核的基因表达和染色质可及性,重建了渗透胁迫下控制根尖发育的TRNs。与在细胞类型水平上常用的计算整合相比,该结果捕获了12,968个峰-基因链接,更准确地重建细胞状态转变过程中基因表达和染色质状态的协调变化。在根尖发育过程中,初始细胞的染色质可及性先于基因表达,这表明染色质可及性的变化可能为细胞的后续分化步骤做好准备。拟时序轨迹分析表明,渗透胁迫可以改变毛原细胞的功能分化。候选的应激相关基因连锁顺式调控元件(gl-cCREs)以及潜在的靶基因也被鉴定出来,并揭示了渗透胁迫下细胞的巨大异质性。
「 技术路线 」
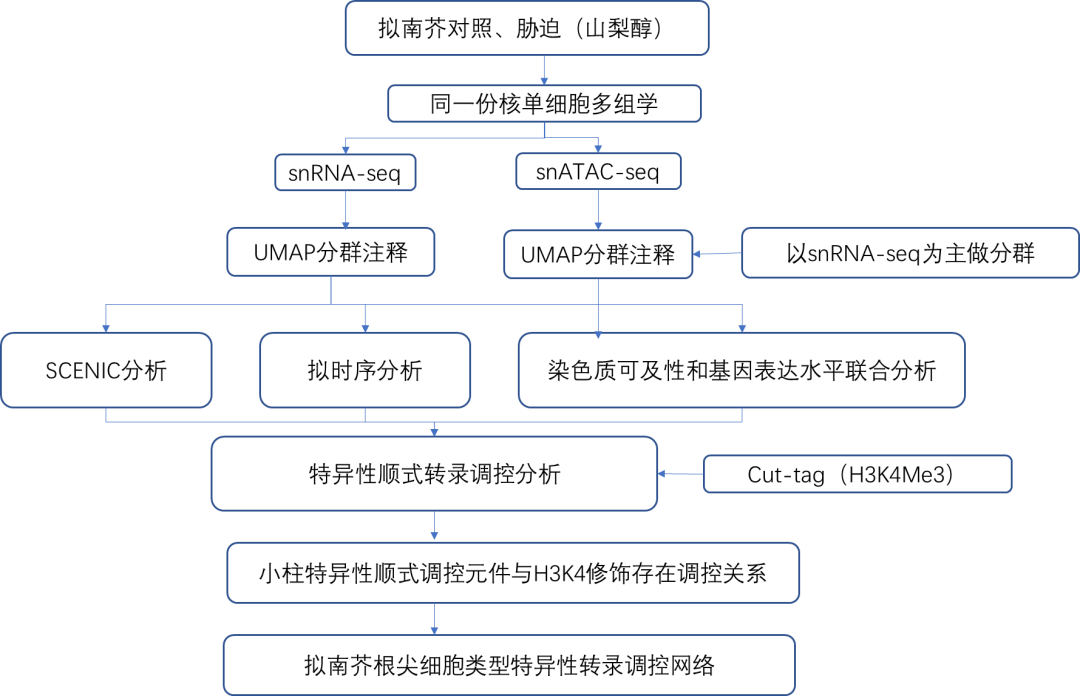
「 研究结果 」
01
单细胞表达谱和单细胞染色质可及性图谱
为了在单细胞水平上研究渗透胁迫下拟南芥根尖的转录组学,作者从正常培养条件(1/2MS)和渗透胁迫(1/2MS+250 mM山梨醇)下生长的5 mM根尖样品(10 d)中提取细胞核。在培养基中添加山梨醇会导致拟南芥典型的渗透胁迫表型,如根长变短、气孔关闭、叶片Ca2+增加等。
单细胞测序共捕获了16,670个细胞核(对照组:5,696个;处理组:10,974),基因中位数为2,433;另获得4,782个RNA UMI计数,3,358个峰值和3,865个ATAC UMI计数。两组样本的TSS(转录起始位点)得分都显示出适合后续分析的值。接下来分别对snRNA-seq和snRNA-seq数据进行了UMAP分群,鉴定出15种不同的细胞类型(分别使用snRNA-seq数据;图1B)和7种不同的细胞类型(单独使用snATAC-seq数据;图1C),根据已知标记物分配细胞类型(图1E,F)。随后,使用10x条形码评估转录组学和表观基因组学数据之间的相关性,发现snRNA-seq和snATAC-seq在大多数细胞类型中匹配,表明从相同细胞中获得的数据具有高可靠性(图1D)。
作者发现单独使用ATAC注释不能像以前的研究那样识别罕见的细胞类型(例如木质部和韧皮部;图1 c)。例如,QC(静止中心)和BBM(木质部极中柱周)和XCP2(木质部极中柱鞘)表现出细胞类型受限的表达。使用ATAC注释不能观察到它们的可及性峰在不同细胞类型间的特异性,但可以通过RNA注释(图1G红框)观察到。通过分析15种细胞类型中来自snATAC-seq的基因活性与来自snRNA-seq的基因表达的相关性,作者发现稀有细胞类型的相关性低于其他细胞类型,这可能是由于来自高丰度细胞的ATAC-seq信号掩盖了来自稀有细胞类型的信号。这强调了在同一细胞内同时分析RNA水平和染色质可及性的必要性,以获得更全面和准确的转录和表观遗传景观特征。由于转录组可以更直接地反映基因表达水平,更适合用于细胞类型的注释,因此在后续分析中,利用RNA注释数据来鉴定细胞类型,并通过细胞特异性id (10x条形码)获得相应的染色质可及性数据。
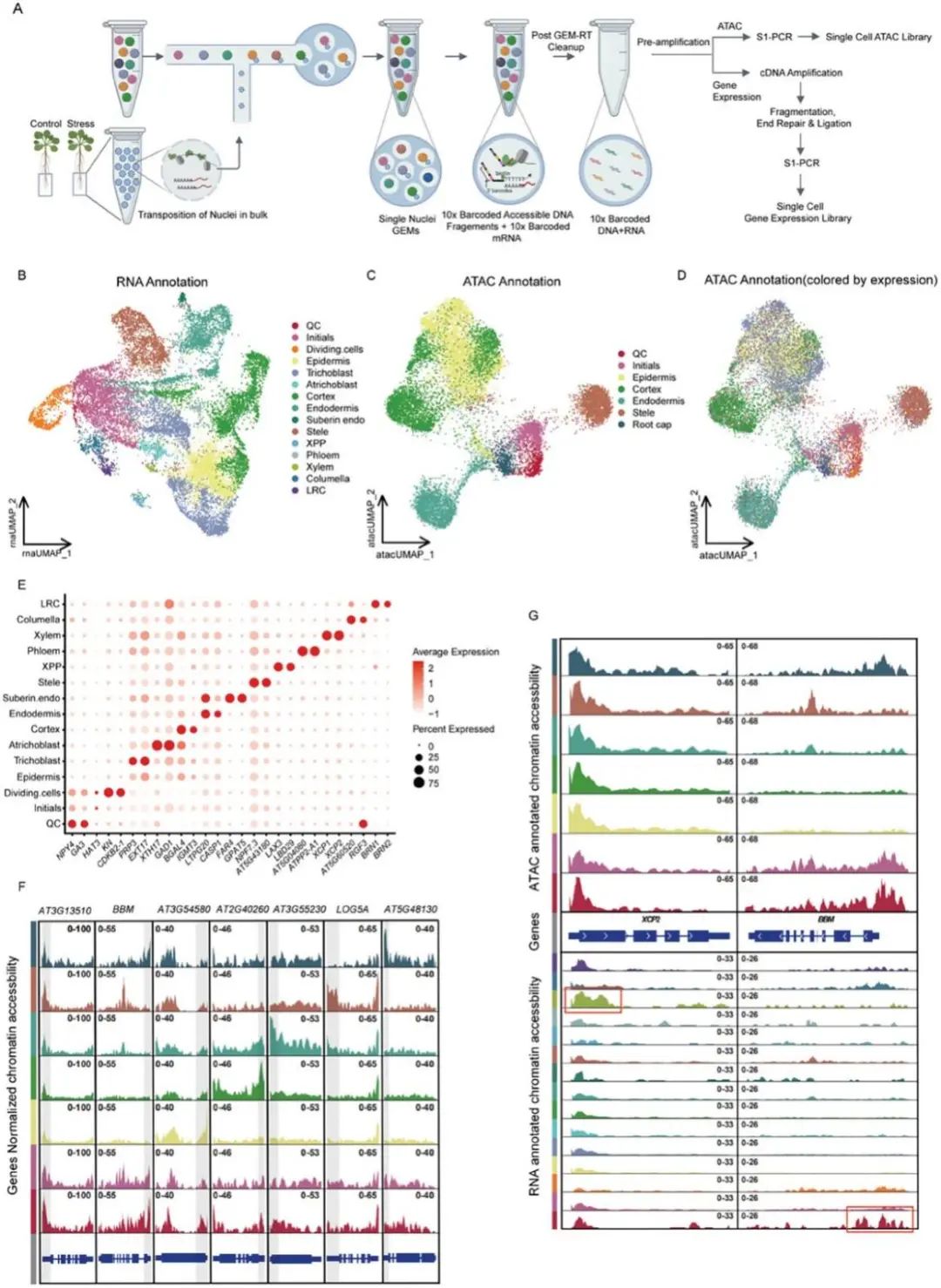
图1:单细胞多个组学产生高质量的全基因组染色质和不同细胞类型的表达谱
02
染色质可及性和基因表达水平差异标记细胞状态
为了直观地观察RNA注释和ATAC注释在每种细胞类型中染色质可及性的差异,分别绘制了联合组、对照组和应激组的桑基图。RNA 序列的桑基图和ATAC-seq相关分析揭示了一个有趣的现象(图2A):大多数初始细胞表现出更开放的染色质区域,类似于分化的细胞(图2A中用黑框和箭头标记)。然而,这些可及基因在初始细胞中的表达水平仍然很低。
利用染色质可及性数据,作者将初始细胞分为6个“亚簇”:INIT.表皮/皮层/内胚层/柱状细胞/根冠(初始细胞表现出更开放的染色质区域,类似于分化的细胞)和INIT.Pr(初始细胞表现出更开放的染色质区域,类似于QC和初始细胞)。大约60.95%(对照)/64.13%(应激)初始细胞表现出与分化细胞相似的染色质可及性。无论是对照还是应激样品中,INIT.表皮/皮质/内胚层/柱状细胞/根冠在染色质状态分类下对应细胞类型的比例相似(图2B)。
此外,作者根据ATAC-seq峰计算了6个“亚簇”的基因得分,并与15种细胞类型进行了相关性分析(图2C)。在对照和应激样本中,INIT.皮层/柱状细胞/内胚层与未来分化的细胞类型相关性最高,而INIT.Pr与其他原代细胞有较高的相关性。在对照样本中,INIT.根冠与小柱的相关性高于LRC(侧根稍),与胁迫结果相反。INIT.表皮与所有最外层细胞的相关性都很高,表皮(对照)与毛原细胞的相关性最高,而表皮(对照)与毛原细胞的相关性最高。表皮(胁迫)与表皮的相关性最高,说明对照的大部分表皮可能发育成毛原细胞,而胁迫的大部分表皮可能发育成表皮。
此外,显微镜观察显示,与对照组相比,渗透培养10天后,毛原细胞的数量减少,这表明渗透胁迫对毛原细胞发育的抑制可以追溯到初始细胞的改变(表现出更开放的染色质区域,类似于表皮细胞),而不仅仅是影响表皮向毛原细胞的分化过程。综上所述,这些结果表明,初始细胞之间染色质可及性的异质性具有一定生物学意义。
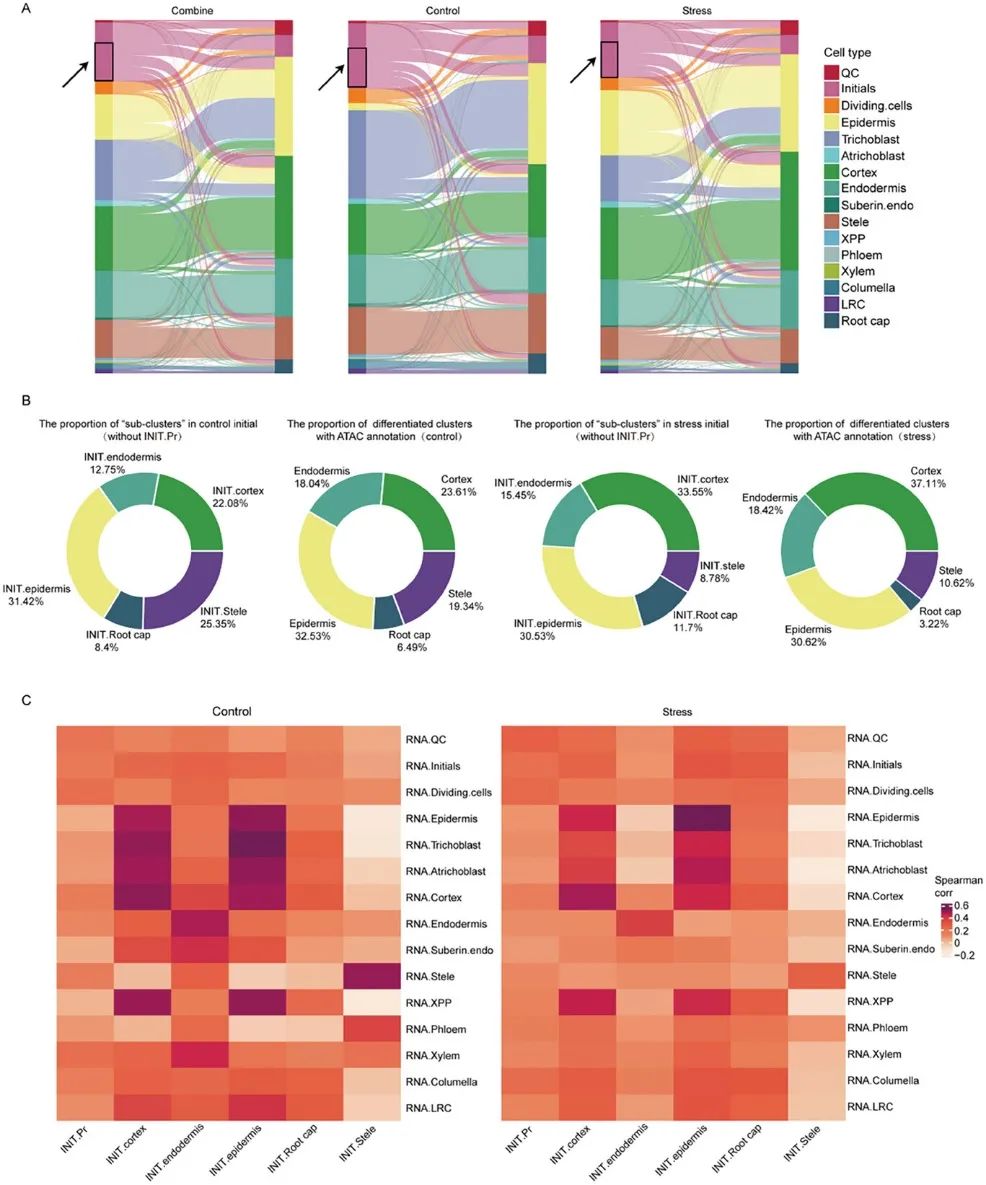
图2:染色质可及性和基因表达水平差异标志着细胞状态
03
原代细胞中染色质可及性预示着未来的细胞命运
作者利用monocle2构建了细胞的初始发育轨迹,并根据染色质可及性分型绘制了细胞的分布图。发育轨迹表明INIT.Pr主要集中在轨迹起始处,与分化细胞染色质可及性相似的细胞位于轨迹尾部(图3A、B);拟时序分析将其发展轨迹划分为三个阶段,并计算了每个阶段中每个“子集群”的比例、INIT.Pr比例在发育初期非常高≈75%,中后期逐渐降低,并伴有分化的“子簇”逐渐增加(图3C,D)。然而,初始标记基因的表达水平在整个发育轨迹中没有显著变化,这表明这些细胞在转录组水平上是相似的,但在随后的分化程序中染色质可及性不同。
为了进一步证实这一推测,分别在对照和应激样本中使用CytoTRACE分析了六个“子簇”的分化顺序。发现在对照组和应激组中INIT.Pr分化明显低于其他“子簇”(图3E)。重建拟时间轨迹的结果表明,在根尖发育过程中,染色质在基因表达开始之前就开启了,可能是为随后的分化步骤做准备,这一现象只能通过测量同一细胞中的染色质状态和基因表达谱来检测。单核多组数据的分析为解释染色质状态和基因表达在细胞类型水平上的不一致提供了新的见解。
在开放的染色质区域中,发现了参与磷吸收的PHT1;1基因,其在根的最外层表皮细胞和一些初始细胞中染色质开放(表现出与表皮相似的染色质可及性)。磷是植物生长发育所必需的大量营养元素之一。增加磷营养水平可以提高植物对干旱的适应能力和水分利用效率。正如预期的那样,PHT1; 1mRNA在最外层表皮细胞中表达,然而,尽管PHT1;1在这些细胞中具有高染色质可及性,但在初始细胞中未表达。作者使用CytoTRACE来追踪QC干细胞、初始细胞、表皮细胞、无壁细胞和毛原细胞的发育轨迹。QC细胞位于轨迹的起点,毛胚细胞位于轨迹的末端(图3F)。在同时分析PHT1;1染色质可及性数据和沿发育轨迹的表达水平后,作者观察到,在一些初始细胞中,染色质为转录起始打开,但该基因未表达(蓝色虚线标记;图3 g, H)。作者还发现,在渗透应激的细胞中,PHT1;1的可达峰数和表达水平高于对照细胞(图3I,J)。
随后,作者重点研究了在拟南芥中已经得到充分研究的渗透胁迫和生长素相关基因,并分析了它们在不同细胞类型中的表达模式和染色质可及性。作者发现,应激组生长素相关基因的基因表达和染色质可及性以细胞类型特异性的方式协调减弱。例如,生长素合成相关基因YUC5在韧皮部被抑制,参与生长素运输的基因ABCB19、PIN1和生长素信号传导相关基因IAA30在QC中被渗透胁迫特异性抑制。此外,应激组渗透胁迫相关基因被细胞型特异性诱导,且可及性峰数高于对照组,即ABA信号通路关键调控因子ABI5在木质部被诱导表达,另一ABA信号转导相关蛋白ABI2在木质部被特异性诱导表达,ABA信号转导相关蛋白ABI2在木质部被特异性诱导表达XPP,提示同一基因家族成员功能分化的空间控制。这些应激相关基因的表达水平仅在一种或两种细胞类型中被特异性诱导,尽管它们的染色质在更多的细胞类型中更容易获得,这表明这些基因的激活受到依赖于细胞类型背景的复杂转录调控的控制。
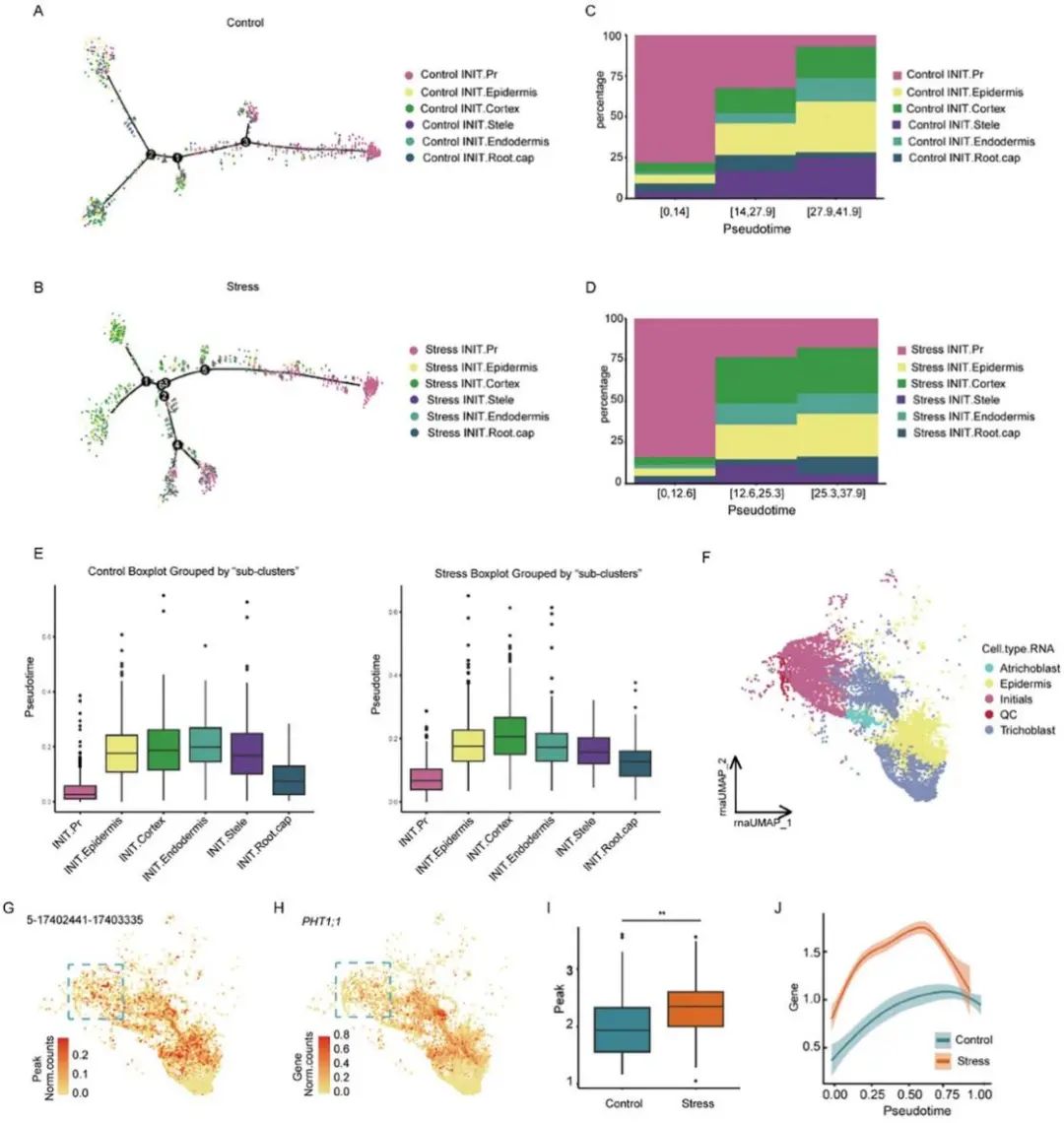
图3:染色质可及性预示着原代细胞未来的细胞命运
04
渗透胁迫下细胞类型特异性变化
渗透胁迫触发基因表达、发育和植物生理的特定反应。利用CytoTRACE对拟南芥根细胞从低分化到高分化的发育轨迹进行了跟踪研究。结合RNA注释结果,作者观察到毛原细胞在尾部富集(表明分化结束),而QC细胞在起始点富集(图4A)。对照细胞比应激细胞相比的具有更大的分化潜力(图4B)。
这些结果表明,渗透胁迫干扰了拟南芥根系中多种细胞类型的生理功能。差异基因表达分析表明,与韧皮部、Suberin-endo和XPP细胞相比,渗透胁迫下毛原细胞、皮层、内胚层和柱状细胞中差异基因表达较多。其中包括编码渗透胁迫相关蛋白激酶的基因,如MPK3和SnRK2.8,这些基因在皮层中被渗透胁迫极大地诱导。在渗透胁迫样本中富集了编码Na+转运体基因HKT1和耐盐基因AT1G13930。观察到另一种蛋白激酶SnRK2.7和根嗜盐相关蛋白SP2L在渗透胁迫下被毛管细胞诱导。上述结果表明,这些细胞类型可能在根尖对渗透胁迫的响应中起着更为关键的作用。GO富集显示除毛原细胞、皮层细胞、内胚层细胞和柱状细胞外,大多数细胞中与水转运和液体转运相关的基因表达水平均升高。
皮层、内皮层和柱状细胞对渗透应激表现出类似的反应,渗透应激诱导这些细胞中与前体代谢物和ATP代谢过程产生相关的基因上调。毛原细胞对渗透胁迫表现出一种特定的反应模式,其基因在有氧呼吸、细胞呼吸和氧化磷酸化相关的途径中富集,这表明每种细胞类型对外部环境因素的反应可能有不同的机制。
毛胚(根毛)是根尖吸收水分、无机盐和感知环境变化的关键细胞类型,与其他细胞类型相比,它在渗透胁迫下的差异表达基因数量最多。通过表型观察,还发现,与对照组相比,应激样品中的毛原细胞减少了,因此,使用CytoTRACE来跟踪毛原细胞的发育轨迹。CytoTRACE结果显示,对照组的毛原细胞分化高于应激组(图4C),与整体水平的结果一致(图4B)。随后,作者分析了渗透胁迫对毛胚发育的影响,利用对照细胞和胁迫细胞重建了毛胚发育的假时间轨迹。作者使用Monocle2对毛原细胞和QC细胞进行拟时间分析,QC被定义为拟时间轨迹中起始位置的起始点(图4D-F)。在起始点附近,应激组和对照组沿同一轨迹发散,在分岔点1形成两个分支:分支1和分支2。
进一步发现,基于整个轨迹的表达变化,基因可以聚为三个簇(图4G)。cluster 1中的基因在Branch 2中高表达,主要调控Branch 2的发育,并伴有缺氧反应相关、铁转运和细胞内铁平衡相关的富集基因,占对照组的很大比例(图4G)。在应激组含有更多毛原细胞的分支1的发育轨迹中,与表皮分化、毛原细胞发育和水分运输相关的基因被高表达(簇2和簇3)(图4G)。拟南芥毛原细胞有两种不同基因表达谱的亚型:一种亚型由成熟的毛原细胞组成,在响应环境变化和离子吸收方面发挥重要作用(Branch2;图4 F);另一个是正在分化的毛胚细胞(Branch1;图4F),其中表达了大多数已验证的毛胚标记基因。以上结果表明,渗透胁迫改变了毛原细胞的生理状态,抑制了部分毛原细胞从分化到成熟的过渡,从而影响了毛原细胞的生理功能(图4D-F)。
植物进化出复杂的转录调控网络(TRNs)来适应干旱和高盐度等渗透胁迫,转录因子(TF)作为分子开关在TRNs中起着核心作用。以往对TF的研究主要集中在整个组织或器官水平,毛原细胞特异性TF调控网络在很大程度上仍未被探索。作者研究了在控制和胁迫条件下的毛胚反调控元件,特别关注了对渗透胁迫的反应。
利用SCENIC分析拟南芥TF家族在毛管细胞中的表达,绘制其调控活性热图和调控网络(图4H,I)。分析发现WRKY17 (AT2G24570)是受非生物胁迫(盐、甘露醇、干旱)诱导的植物特异性WRKY家族TF中的一员,在渗透胁迫下在毛管细胞中高表达(图4H)。与此一致的是,WRKY17的基序可及性在毛胚中增加。更重要的是,进一步观察到启动子含有WRKY17结合基序的靶基因在应激组中表达上调。在毛原细胞调控网络中,也发现了WRKY11(AT4G31550), 它在系统发育上与WRKY17相关,也受渗透胁迫诱导(图4H)。WRKY11的基序可及性和靶基因表达模式与WRKY17相似。利用qPCR检测了WRKY17和WRKY11在对照和渗透处理的拟南芥根尖中的表达水平。作者观察到与对照组相比,渗透处理的样品中WRKY17和WRKY11的表达水平更高。WRKY转录因子在调节植物对生物和非生物胁迫的反应中起着至关重要的作用,尽管对多种植物进行了研究,但WRKY转录因子对非生物胁迫的细胞类型特异性仍不完全清楚。通过单细胞多组学技术发现WRKY11/17在胁迫下的毛管细胞中高表达,并可能在渗透胁迫响应中发挥关键作用。
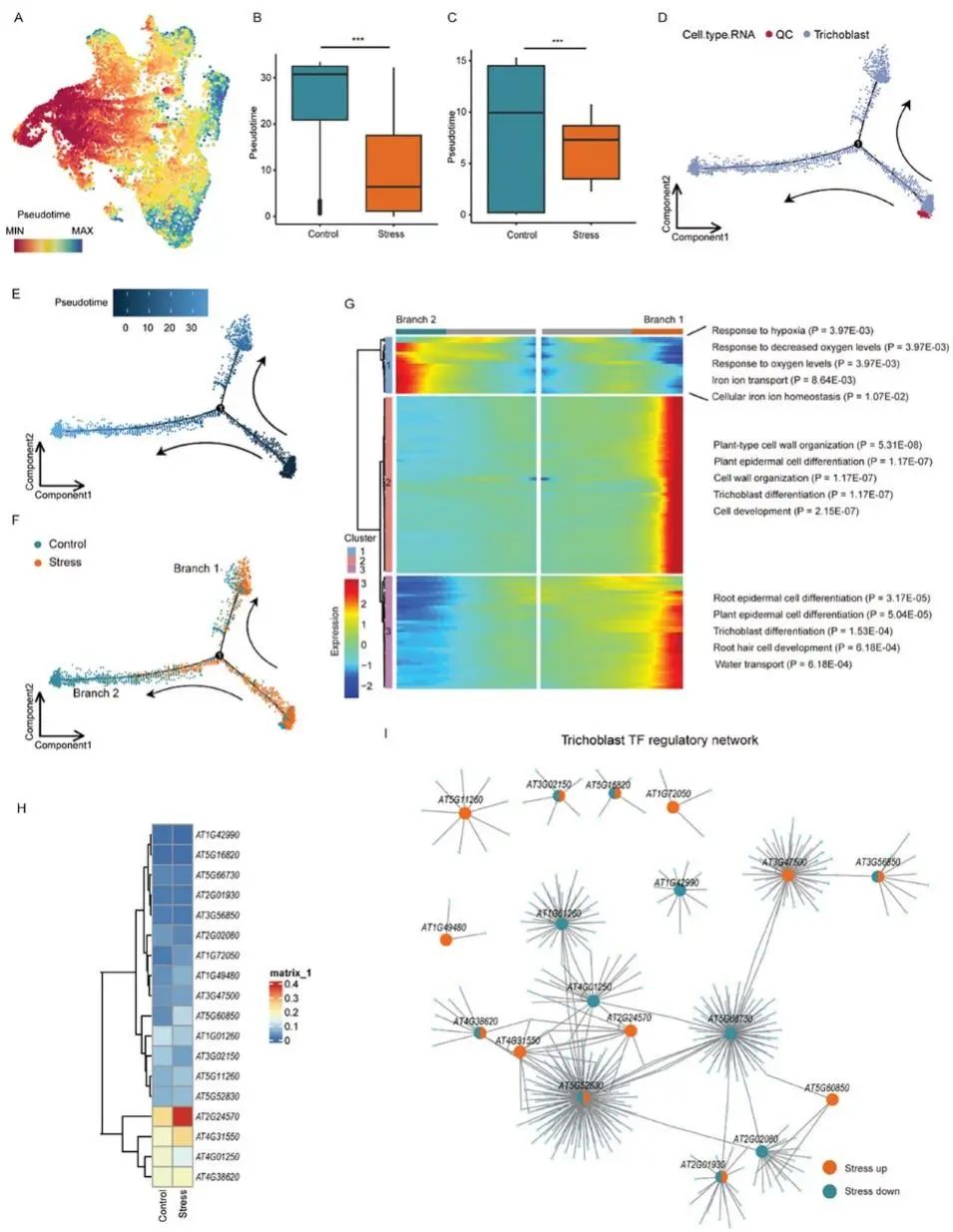
图4:渗透胁迫下细胞类型特异性变化
05
渗透胁迫下细胞类型特异性顺式转录调控
植物中的基因表达受顺式调控元件(CRE)调控,如启动子和增强子,它们响应发育和环境变化。虽然在组织水平上对拟南芥CRE的研究已经取得了实质性进展,但使用单细胞多组学技术可以更深入地了解细胞类型特异性基因调控程序。通过在同一细胞中使用单细胞多组ATAC +基因表达,旨在以更高的准确性鉴定细胞特异性基因连锁候选顺式调控元件(gl-cCREs)。通过分析snATAC-seq数据检测共可及性峰对来预测基因组中的顺式调控相互作用,其中一个峰位于启动子区域,染色质可及性与下游基因表达呈正相关。因此,作者预测了一组CRE启动子对,并在伪批量中鉴定了7373个gl-cCREs和3763个cCRE连锁基因。并发现验证的拟南芥根增强子序列与gl-cCREs之间存在重叠,证实了数据的可靠性。
随后利用snATAC-seq数据预测不同细胞类型中与应力相关的gl-cCREs,发现基于倍数变化直方图,与应力相关的gl-cCREs数量远高于假体,小柱应力样品中与应力相关的gl-cCREs数量比根状茎高约10倍(图5A)。这些结果表明,单细胞多组ATAC +基因表达能够获得更全面的应激cCREs信息,因为在snRNA-seq和sATAC-seq数据的细胞类型水平整合下,一些细胞特异性峰被淹没,因此无法识别。
然后,重点分析了具有比异常值更多相互作用峰对的应力相关gl-cCREs,将其定义为应力相关热点gl-cCREs (hgl- cCREs)。共发现240例hglcCREs(图5B-F)。先前的研究表明,增强子在不同物种中具有相似的功能,并驱动发育基因的细胞特异性表达。为了探索hgl-cCREs的保守性,分别将gl-cCREs和hgl-cCREs序列与已发表的十字花科保守非编码序列进行了比较。发现大多数hgl-cCREs含有多个保守的非编码序列,并且hgl-cCREs含有这些序列的比例明显高于gl-cCREs(图5G)。这些结果表明,一些保守的非编码序列可能作为增强元件调节拟南芥在渗透胁迫下多个基因的表达(图5H),这些序列可能在其他十字花科物种中也有类似的作用,如油菜和阿拉伯蒿。
鉴于小柱细胞中与应激相关的gl-cCREs和靶基因的数量明显高于其他细胞类型(图5A,I),对应激相关的gl-cCREs靶基因进行富集分析。富集分析表明,这些基因与组蛋白H3K4甲基化相关,要么是组蛋白H3K4甲基化复合物的核心组分,要么参与组蛋白H3K4甲基化和其他关键生物学过程的调控(图5J)。顺式调控元件分析表明,胁迫相关的gl-cCREs主要参与激活小柱渗透胁迫下H3K4甲基化相关基因的表达。同时,检测了组蛋白甲基化在渗透胁迫下的变化,并使用CUT&Tag技术检测了对照组和应激组中H3K4me3的总体水平。测序数据表明,在渗透胁迫下修饰水平显著增加。对H3K4me3修饰基因的富集分析显示,渗透胁迫下参与氧化磷酸化、铁转运、茉莉酸合成和损伤反应的H3K4me3相关基因上调。小柱特异性顺式调控元件与H3K4修饰之间可能确实存在调控关系;然而,确切的机制需要更详细的研究。
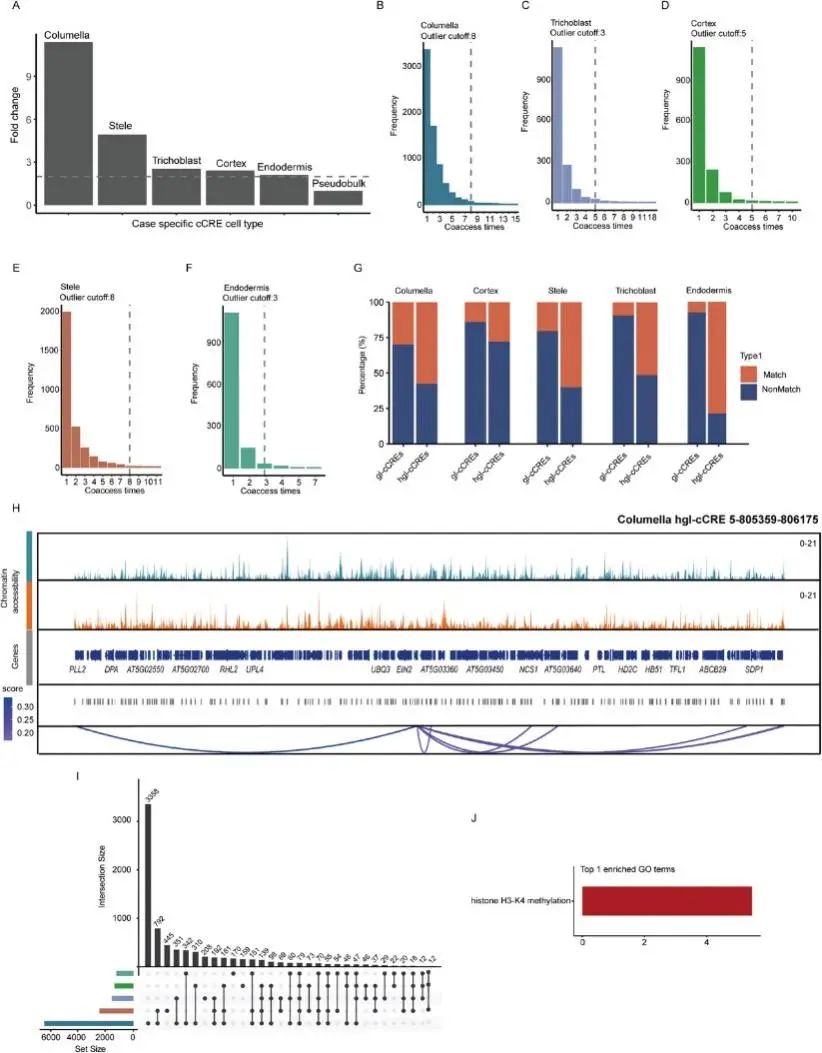
图5:渗透胁迫下细胞型特异性顺式转录调控
目前,单细胞测序正当火热,结合单细胞ATAC和单细胞转录组数据更能帮助我们挖掘细胞异质性基因的调控关系。通过表观基因组学和基因表达谱的综合分析来鉴定复杂的细胞群体,并捕获细胞异质性,从而发现隐藏的信息,利用基因表达标志物解释表观遗传图谱,发现新的基因调控作用,探索新的调控机制。
















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









