






-
富集分析:(五)clusterProfiler:Visualization | 生信技工 (yanzhongsino.github.io)
-
保存的clusterProfiler富集分析结果做可视化
- 如果是clusterProfiler的enrichGO(),gseGO(),enricher(),gseGO()等函数的结果
ego保存成的文件,已关闭R环境。 - 可导入文件,新建enrichResult对象ego,再进行下一步可视化。
- 这里假设用R命令
write.table(as.data.frame(ego),"go_enrich.csv",sep="\t",row.names =F,quote=F)保存ego在go_enrich.csv文件。1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18
data<-read.table("go_enrich.csv",sep="\t",header=T,quote="") head(data,2) #查看data前2行 ONTOLOGY ID Description GeneRatio 1 BP GO:0010222 stem vascular tissue pattern formation 12/349 2 BP GO:0010588 cotyledon vascular tissue pattern formation 12/349 BgRatio pvalue p.adjust qvalue 1 29/16975 1.792157e-13 2.107577e-10 1.790270e-10 2 39/16975 1.122611e-11 6.600951e-09 5.607145e-09 geneID 1 mc11300/mc11301/mc19080/mc19081/mc26300/mc31693/mc37850/mc40780/mc40781/mc40782/mc40784/mc40918 2 mc11300/mc11301/mc19080/mc19081/mc26300/mc31693/mc37850/mc40780/mc40781/mc40782/mc40784/mc40918 Count 1 12 2 12 geneID_all <- unlist(apply(as.matrix(data$geneID),1,function(x) unlist(strsplit(x,'/')))) #得到富集到的所用geneID ego<-new("enrichResult", result=data, gene=geneID_all, pvalueCutoff=0.01,pAdjustMethod="BH",qvalueCutoff=0.05,ontology="BP",keytype="GID",universe='Unknown',geneSets=list(),organism="Unknown",readable=FALSE) #把data内容赋值给ego的result,geneID_all赋值给gene,每个富集到的GO对应的gene集应该赋值给geneSets(数据是字典(键值对是GOID和geneIDs)组成的列表,这里直接给了空的),ontology与enrichGO分析的ont参数一致,这里的pvalueCutoff=0.01,pAdjustMethod="BH",qvalueCutoff=0.05根据富集分析参数的设置,或者随意设置或者不设置也不会影响可视化。
- 其他来源富集分析结果可视化
如果是其他软件的富集分析结果,可以根据ego的result变量格式进行修改格式,改成go_enrich.csv相同的格式的文件,再按照上面的步骤导入文件,并保存到新建的ego对象。即可用clusterProfiler的可视化包可视化其他软件的富集分析结果了。
2. 功能富集结果可视化
下面的可视化大多基于在R中已获得富集分析的结果ego。
2.1. enrichplot包
enrichplot包有几种可视化方法来解释富集结果,支持clusterProfiler获得的ORA和GSEA富集结果。
2.1.1. 安装和载入
安装和载入enrichplot包
1 2 | BiocManager::install("enrichplot")
library(enrichplot)
|
2.1.2. 可视化包
- 推荐dotplot或barplot可视化前10个GO Terms条目。
- 推荐goplot有向无环图查看富集的GO Terms间的关系。
- 可视化barplot —— 条形图
将富集分数(例如p 值)和基因计数或比率描述为条形高度和颜色。横轴为该GO term下的差异基因个数,纵轴为富集到的GO Terms的描述信息, showCategory指定展示的GO Terms的个数为20个,默认展示显著富集的top10个,即p.adjust最小的10个。
barplot(ego, showCategory=20, title="EnrichmentGO_MF")
使用mutate导出的其他变量也可以用作条形高度或颜色。
1 2 | mutate(ego, qscore = -log(p.adjust, base=10)) %>%
barplot(x="qscore")
|

Figure 1. Bar plot of enriched terms
from clusterProfiler book
- 可视化dotplot —— 点阵图
dotplot(edo, showCategory=30) + ggtitle("dotplot for ORA")
dotplot(edo2, showCategory=30) + ggtitle("dotplot for GSEA")
散点图,横坐标为GeneRatio,纵坐标为富集到的GO Terms的描述信息,showCategory指定展示的GO Terms的个数,默认展示显著富集的top10个,即p.adjust最小的10个。
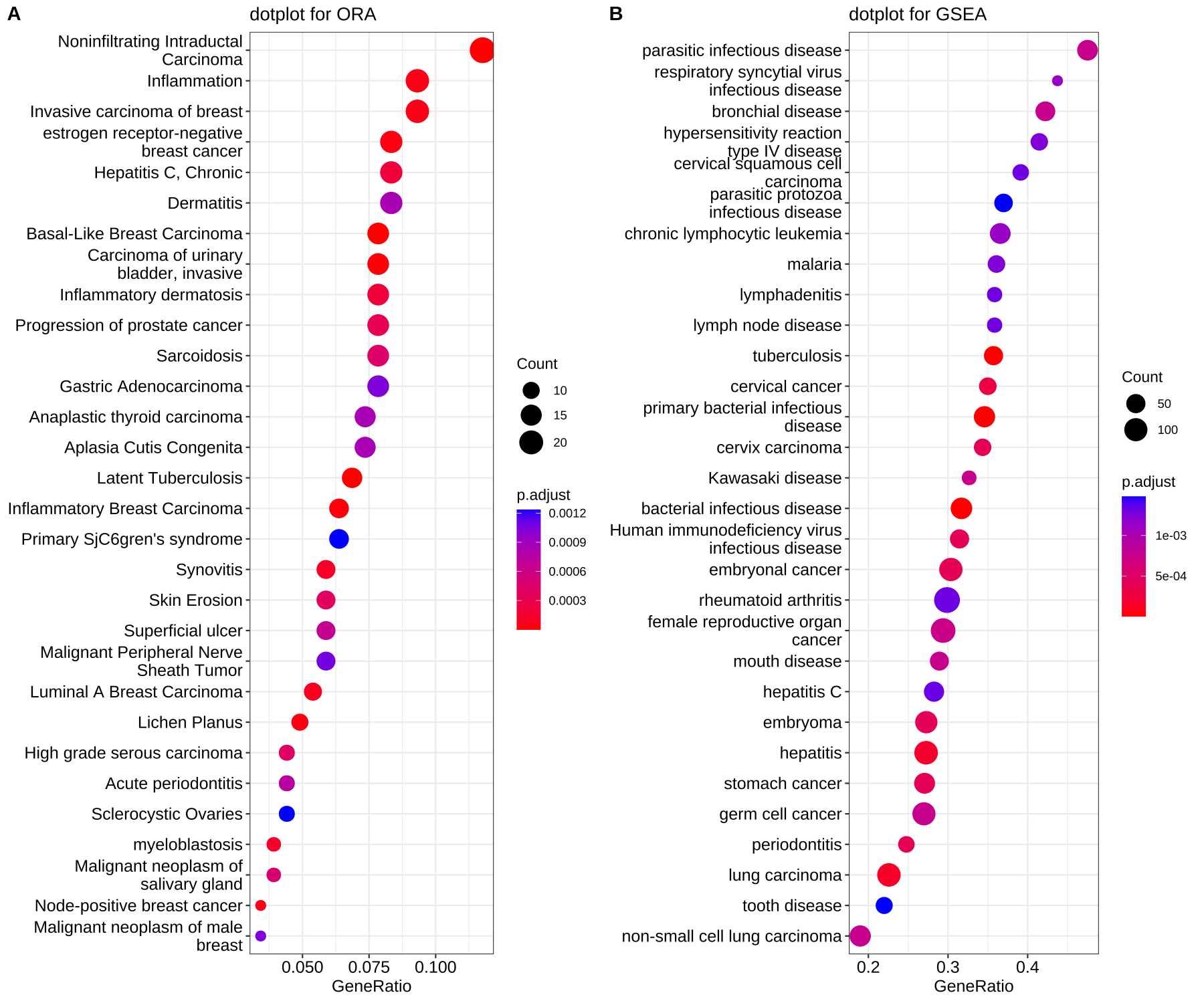
Figure 2. Dot plot of enriched terms
from clusterProfiler book
- 可视化cnetplot —— 类别网络图
cnetplot 将基因和生物学概念(例如 GO 术语或 KEGG 通路)的联系描述为一个网络(有助于查看哪些基因涉及富集通路和可能属于多个注释类别的基因)。对于基因和富集的GO terms之间的对应关系进行展示,如果一个基因位于一个GO Terms下,则将该基因与GO连线。图中灰色的点代表基因,黄色的点代表富集到的GO terms, 默认画top5富集到的GO terms, GO 节点的大小对应富集到的基因个数。
cnetplot(ego, categorySize = "pvalue", foldChange = gene_list
1 2 3 4 5 6 7 | ## convert gene ID to Symbol edox <- setReadable(ego, 'org.Hs.eg.db', 'ENTREZID') p1 <- cnetplot(edox, foldChange=geneList) ## categorySize can be scaled by 'pvalue' or 'geneNum' p2 <- cnetplot(edox, categorySize="pvalue", foldChange=geneList) p3 <- cnetplot(edox, foldChange=geneList, circular = TRUE, colorEdge = TRUE) cowplot::plot_grid(p1, p2, p3, ncol=3, labels=LETTERS[1:3], rel_widths=c(.8, .8, 1.2)) |
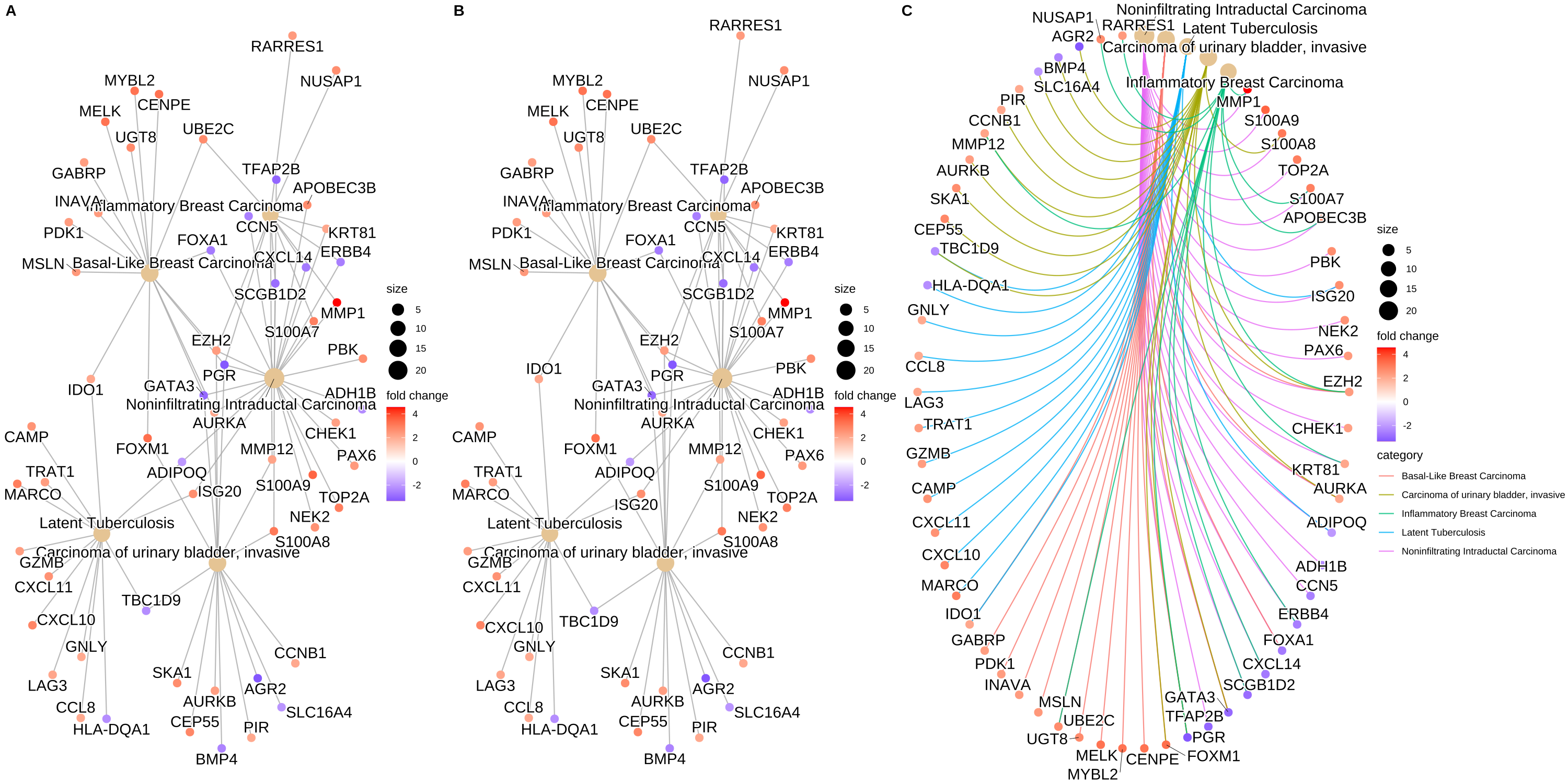
Figure 3. Network plot of enriched terms
from clusterProfiler book
- 可视化heatplot —— 类热图功能分类
同样使用edox。
heatplot类似cnetplot,而显示为热图的关系。
如果用户想要显示大量重要术语,那么类别网络图可能会过于复杂。在heatplot能够简化结果和更容易识别的表达模式。
1 2 3 | p1 <- heatplot(edox, showCategory=5) p2 <- heatplot(edox, foldChange=geneList, showCategory=5) cowplot::plot_grid(p1, p2, ncol=1, labels=LETTERS[1:2]) |
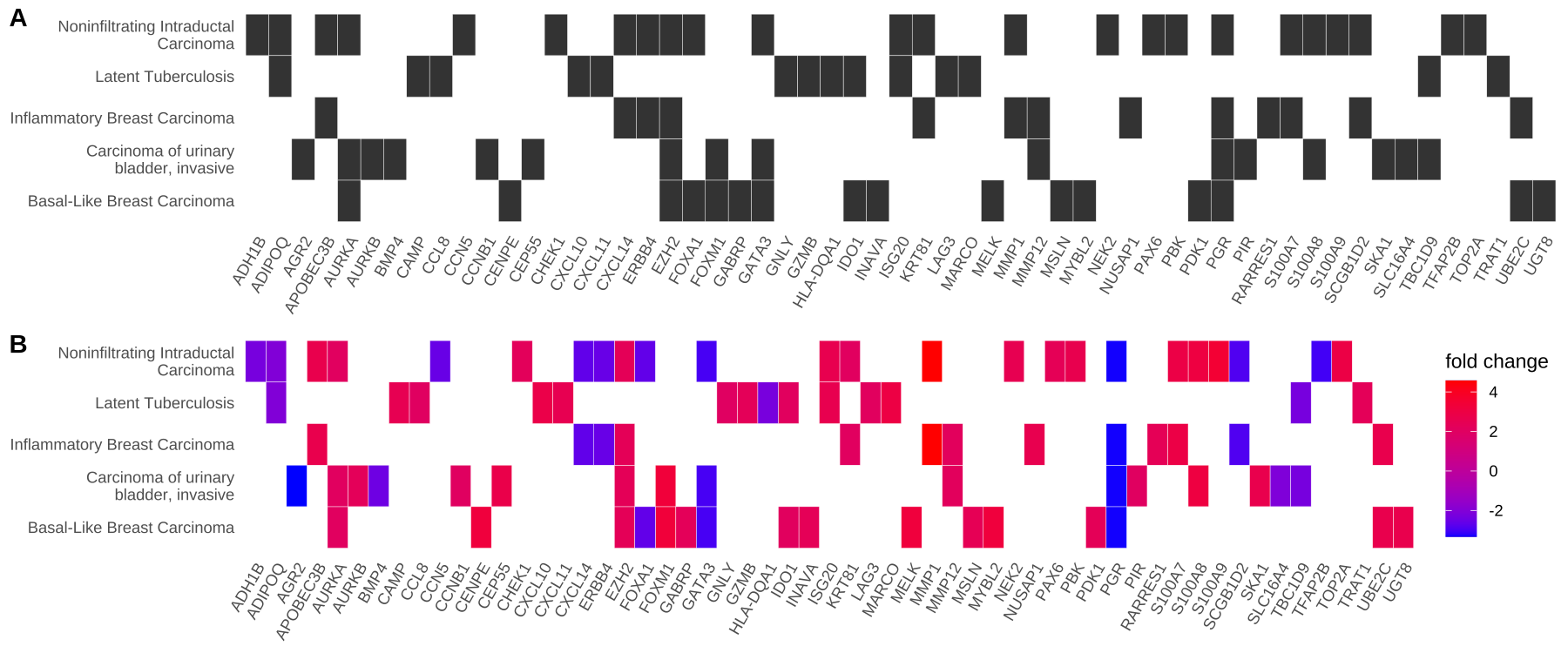
Figure 4. Heatmap plot of enriched terms
from clusterProfiler book
- 可视化treeplot —— 树状图
treeplot()函数执行丰富术语的层次聚类。它依赖于pairwise_termsim()函数计算的丰富项的成对相似性,默认情况下使用 Jaccard 的相似性指数 (JC)。如果支持,用户还可以使用语义相似度值(例如,GO、DO和MeSH)。
默认聚合方法treeplot()是ward.D,用户可以通过hclust_method参数指定其他方法(例如,’average’、’complete’、’median’、’centroid’等。
treeplot()函数会将树切割成几个子树(由nCluster参数指定(默认为 5))并使用高频词标记子树。
1 2 3 4 | edox2 <- pairwise_termsim(edox) p1 <- treeplot(edox2) p2 <- treeplot(edox2, hclust_method = "average") aplot::plot_list(p1, p2, tag_levels='A') |

Figure 5. Tree plot of enriched terms
from clusterProfiler book
- 可视化emapplot —— 富集图
对于富集到的GO terms之间的基因重叠关系进行展示,如果两个GO terms系的差异基因存在重叠,说明这两个节点存在overlap关系,在图中用线条连接起来。每个节点是一个富集到的GO term, 默认画top30个富集到的GO terms, 节点大小对应该GO terms下富集到的差异基因个数,节点的颜色对应p.adjust的值,从小到大,对应蓝色到红色。
1 2 3 4 5 6 | ego2 <- pairwise_termsim(ego) p1 <- emapplot(ego2) p2 <- emapplot(ego2, cex_category=1.5) p3 <- emapplot(ego2, layout="kk") p4 <- emapplot(ego2, cex_category=1.5,layout="kk") cowplot::plot_grid(p1, p2, p3, p4, ncol=2, labels=LETTERS[1:4]) |
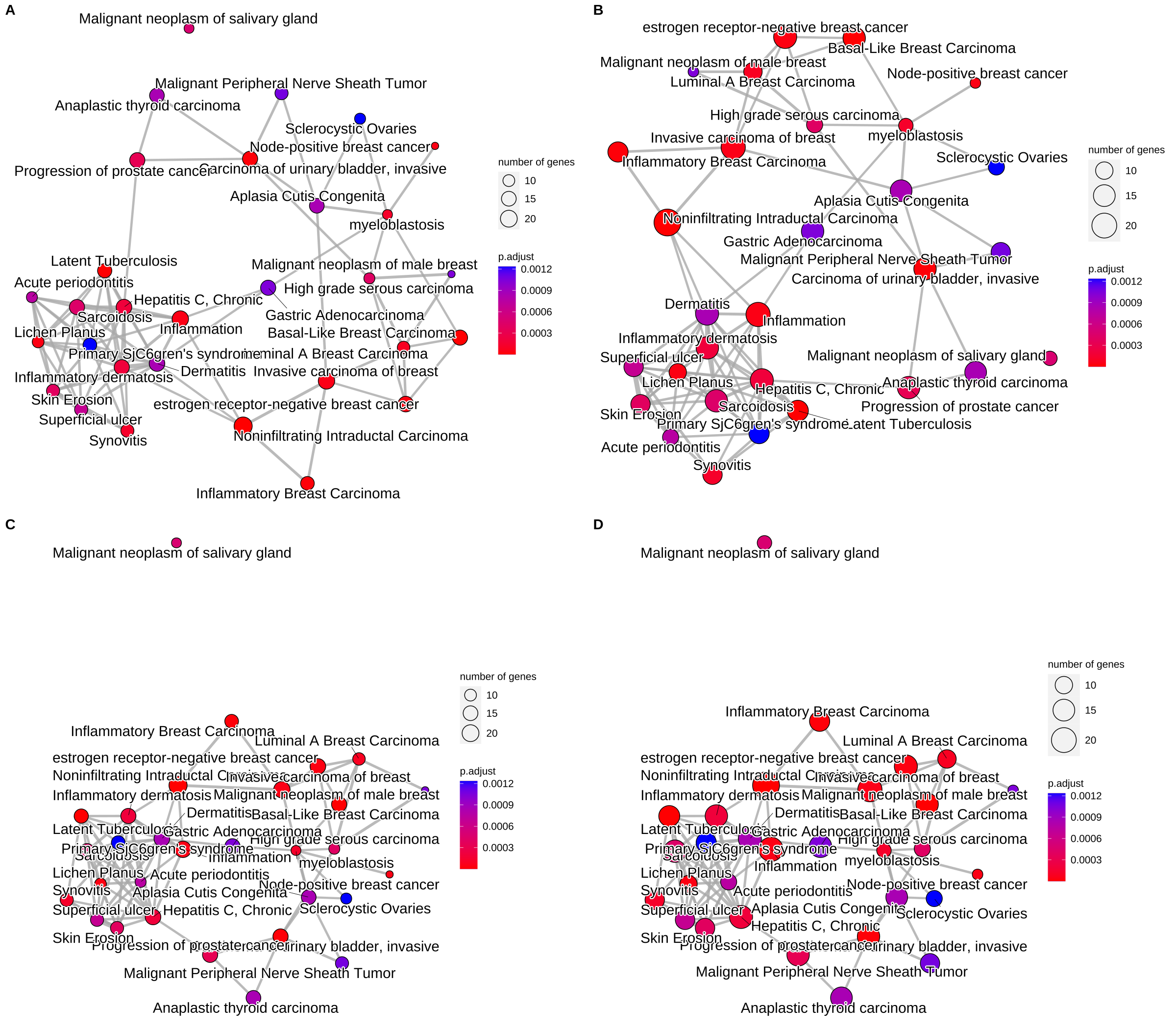
Figure 6. Plot for results obtained from hypergeometric test and gene set enrichment analysis. default (A), cex_category=1.5 (B), layout=”kk” (C) and cex_category=1.5,layout=”kk” (D).
from clusterProfiler book
- 可视化upsetplot —— upset图
upsetplot是cnetplot可视化基因和基因集之间复杂关联的替代方法。它强调不同基因集之间的基因重叠。
upsetplot(ego)
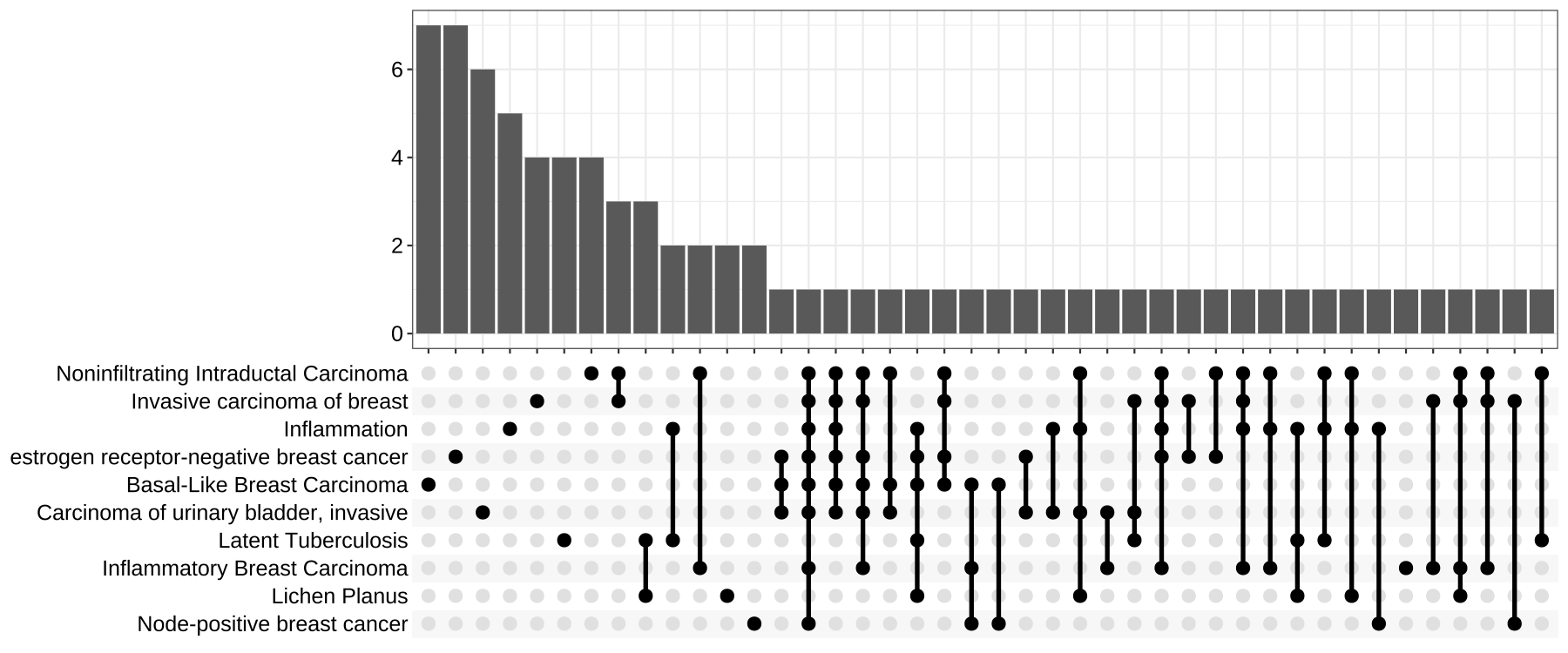
Figure 7. Upsetplot for over-representation analysis.
from clusterProfiler book
- 可视化ridgeplot —— 脊线图
ridgeplot将可视化核心富集基因的表达分布为GSEA富集类别。它帮助用户解释上调/下调的途径。
ridgeplot(ego)
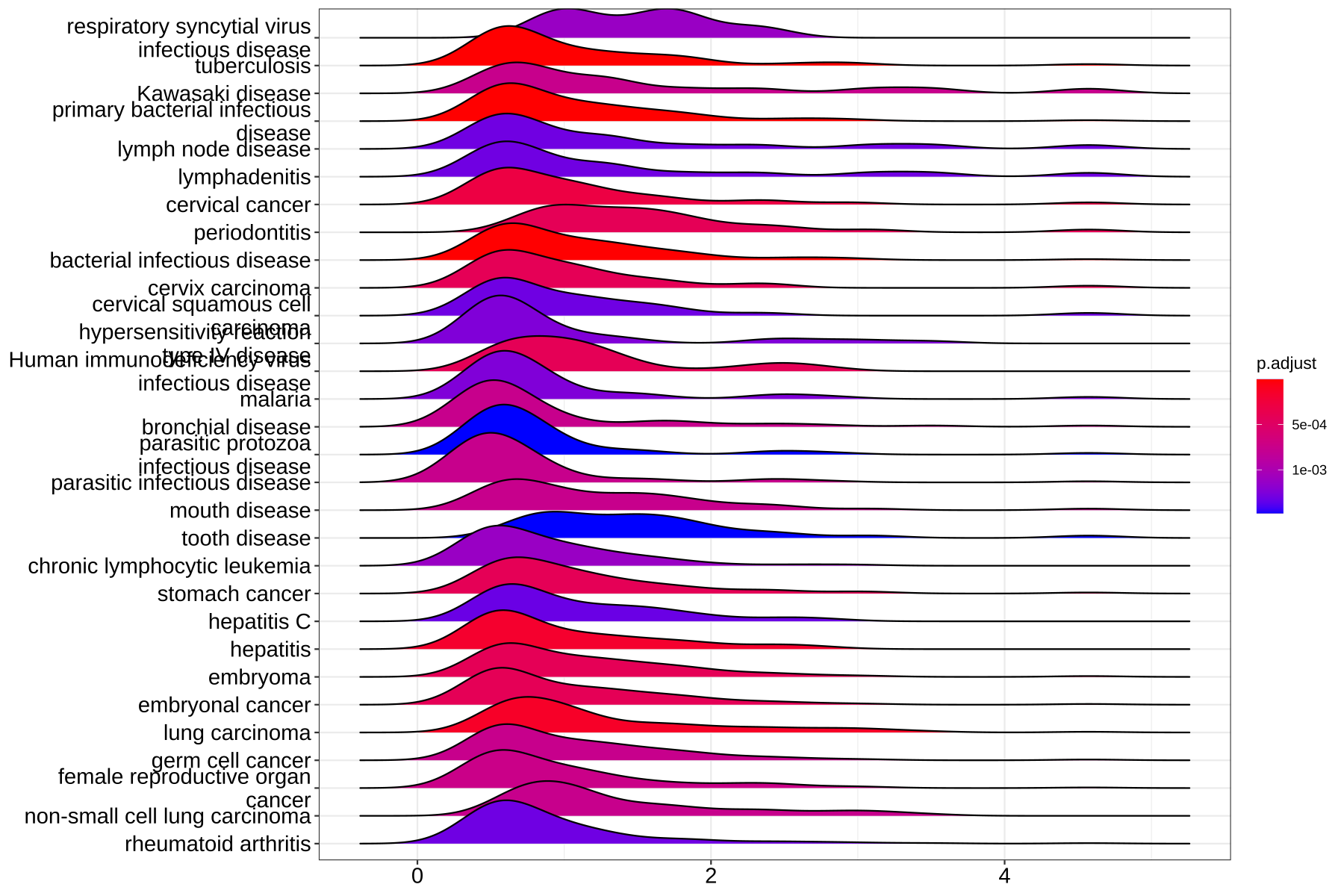
Figure 8. Ridgeplot for gene set enrichment analysis.
from clusterProfiler book
2.2. 可视化plotGOgraph/goplot —— 有向无环图
plotGOgraph(ego, firstSigNodes=10)
- 有向无环图(Directed acyclic graph, DAG),矩形代表富集到的top10个GO Terms,颜色从黄到红,对应p值从大到小。和topGO做富集分析的DAG图一样。
当enrichGO富集分析时ont参数选了ALL时,结果文件会在第一列前增加一列ONTOLOGY为子类,这时直接用于plotGOgraph画图会报错。
试了下,下面两种方案还是会报错Error in if (!ont %in% c(“BP”, “MF”, “CC”)) { :argument is of length zero。。还是尽量在enrichGO分析时就用ont=”BP”吧。
- 可以在结果文件中筛选出特定子类(比如BP)的结果行,并删除第一列ONTOLOGY后保存文件,再读进R用于plotGOgraph画图。
- 也可以在R内用命令
ego2<-ego%>%filter(ONTOLOGY== "BP")筛选BP子类,接着用ego3<-ego2%>%select(!ONTOLOGY)或者ego3<-ego2[,-1]删除第一列(即ONTOLOGY列),然后用plotGOgraph(ego3)作图。
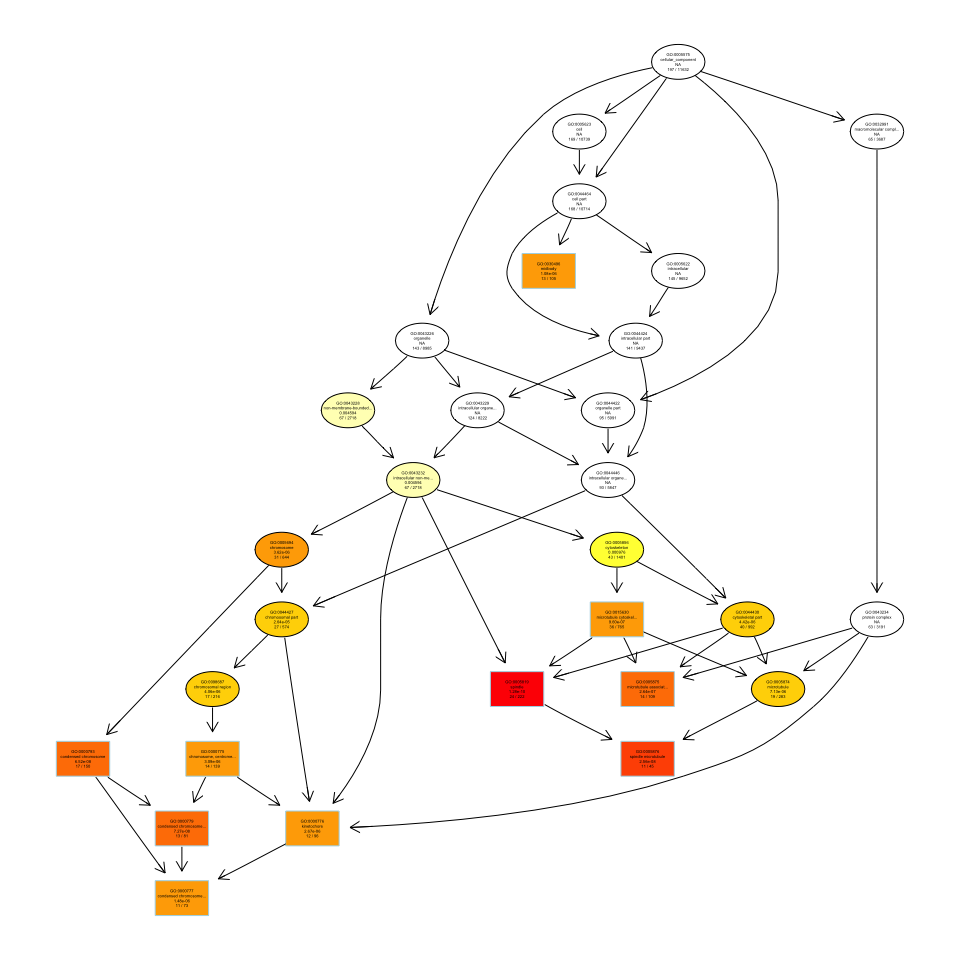
Figure 9. DAG图
from clusterProfiler blog
goplot(ego, showCategory = 10)
- igraph布局方式的有向无环图
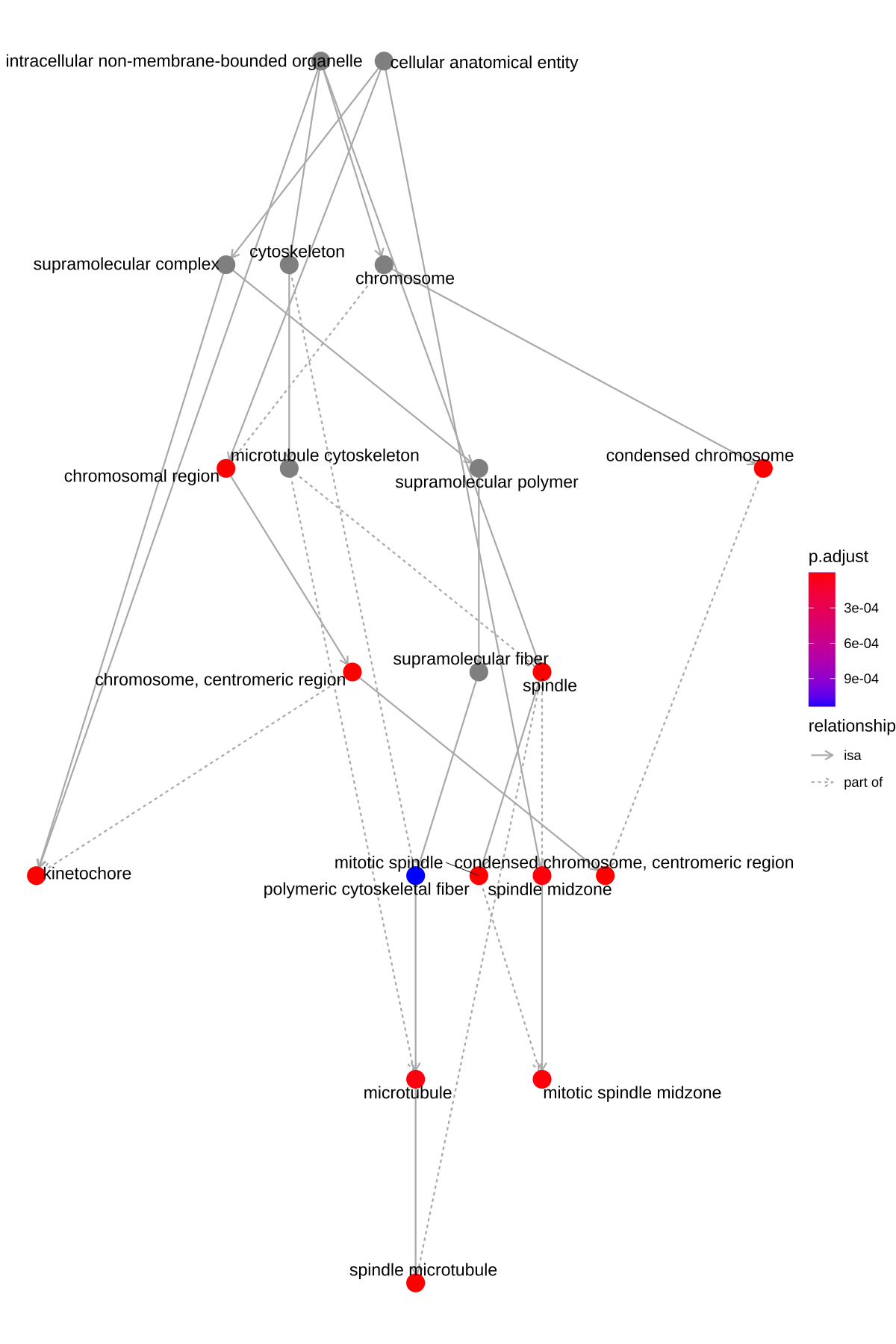
Figure 10. goplot的DAG图
from clusterProfiler book
2.3. 可视化 —— wordcloud
词云的方式显示结果
1 2 3 4 5 | install.packages("wordcloud")
library(wordcloud)
wcdf <- read.table(text = ego$GeneRatio, sep = "/")[1]
wcdf$term <- ego[,2]
wordcloud(words = wcdf$term, freq = wcdf$V1, scale=(c(4, .1)), colors=brewer.pal(8, "Dark2"), max.words = 25)
|

Figure 11. wordcloud词云图
from NGS Analysis ebook
3. 导出可视化结果
- Rstudio
如果是在Rstudio中,可以直接看到绘图结果,导出需要的文件格式即可。
- 代码导出
1 2 3 4
pdf("ego.pdf") #如果保存png,就改成png("ego.png") ego_fig<-barplot(x) #画图函数 print(ego_fig) #画到pdf文件 dev.off() #关闭pdf画板



















 2324
2324

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











