






完成了前面的基础质控、过滤以及去除细胞周期的影响后,我们可以开始SCTransform normalization。
SCTransform normalization的优势:
- 1️⃣ 一个
SCTransform函数即可替代NormalizeData,ScaleData,FindVariableFeatures三个函数;- 2️⃣ 对测序深度的校正效果要好于
log标准化(10万以内的细胞都建议使用SCT);- 3️⃣
SCTransform,可用于矫正线粒体、细胞周期等因素的影响,但不能用于批次矫正;- 4️⃣ 改善信/噪比;
- 5️⃣ 发现稀有细胞。
用到的包
rm(list = ls())
library(Seurat)
library(tidyverse)
library(SingleR)
library(celldex)
library(RColorBrewer)
library(SingleCellExperiment)
library(ggsci)
示例数据
这里我们还是使用之前建好的srat文件,我之前保存成了.Rdata,这里就直接加载了。
load("./srat1.Rdata")
srat
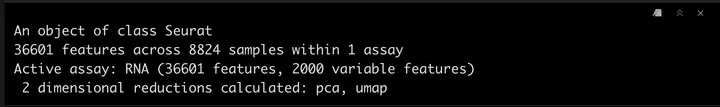
计算细胞周期评分
4.1 新版基因集
这次我们用新版的细胞周期基因集。
cc.genes.updated.2019

4.2 计算评分
s.genes <- cc.genes.updated.2019$s.genes
g2m.genes <- cc.genes.updated.2019$g2m.genes
srat <- CellCycleScoring(srat, s.features = s.genes, g2m.features = g2m.genes)
table(srat[[]]$Phase)

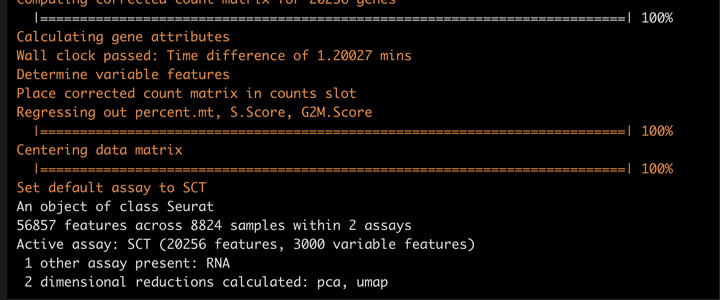
SCTransform normalization
这里我们用一个函数就可以完成。
srat <- SCTransform(srat,
method = "glmGamPoi",
ncells = 8824,
vars.to.regress = c("percent.mt","S.Score","G2M.Score"),
verbose = T)
srat
降维与聚类
我们这里进行一下的标准降维、聚类,这里的dims推荐大家尽可能设置的大一些。
srat <- RunPCA(srat, verbose = F)
srat <- RunUMAP(srat, dims = 1:30, verbose = F)
srat <- FindNeighbors(srat, dims = 1:30, verbose = F)
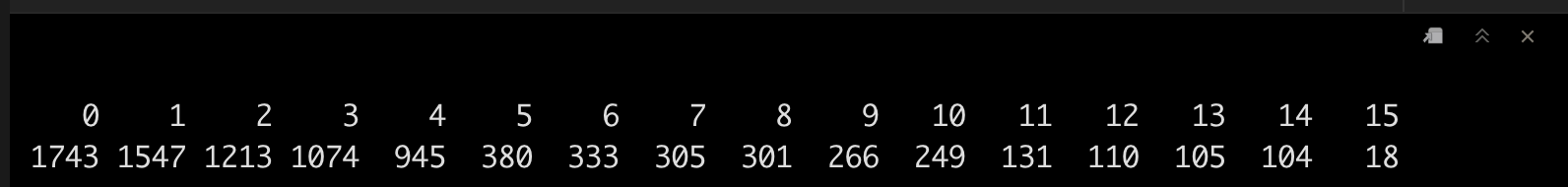
srat <- FindClusters(srat, verbose = F)
table(srat[[]]$seurat_clusters)
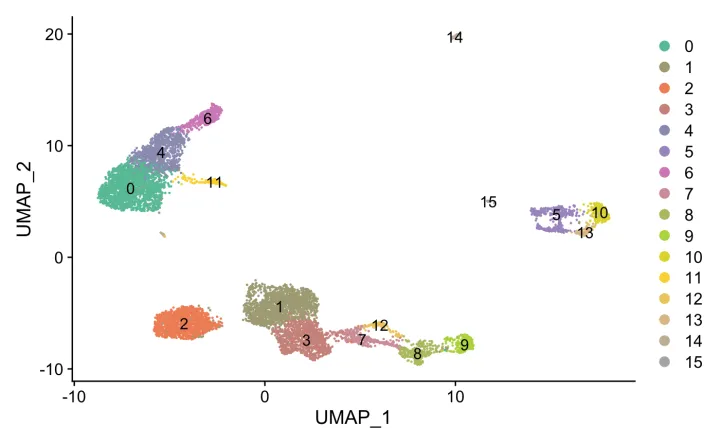
可视化一下吧。
ncluster <- length(unique(srat[[]]$seurat_clusters))
mycol <- colorRampPalette(brewer.pal(8, "Set2"))(ncluster)
DimPlot(srat, label = T,
cols = mycol)
稀有细胞marker探索
接着我们探索一下血小板和树突状细胞的marker,分别为PPBP和LILRA4。
7.1 可视化
这里我们可以发现在PPBP在一个极小的细胞群中没有被标注出来。
FeaturePlot(srat,c("PPBP","LILRA4"),
label = T,
cols = colorRampPalette(brewer.pal(11, "Spectral"))(10)
)
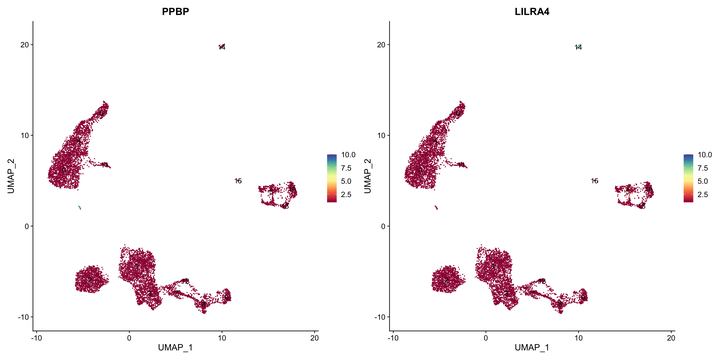
7.2 解决方案
我们可以通过提高FindClusters函数中的resolution选项来提高聚类数量。
当然最简答的办法就是手动标记啦,这里就不演示啦。
srat <- FindNeighbors(srat, dims = 1:30, k.param = 15, verbose = F)
## Leiden algorithm即algorithm = 4, 需要配置python环境
srat <- FindClusters(srat, verbose = F, algorithm = 4, resolution = 0.95)
看一下现在有多少个聚类吧。
table(srat[[]]$seurat_clusters)

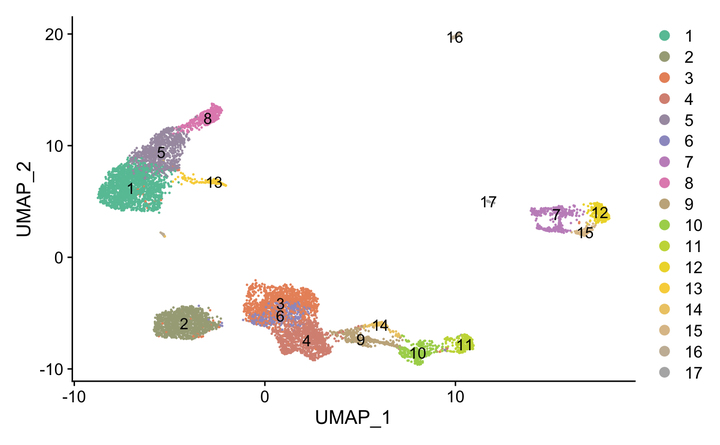
可视化一下吧!~
ncluster <- length(unique(srat[[]]$seurat_clusters))
mycol <- colorRampPalette(brewer.pal(8, "Set2"))(ncluster)
DimPlot(srat, label = T,
cols = mycol)



















 1053
1053

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











