






之前,我们已经介绍过其他的整合方法:
今天来看看Cell发表的LIGER

介绍
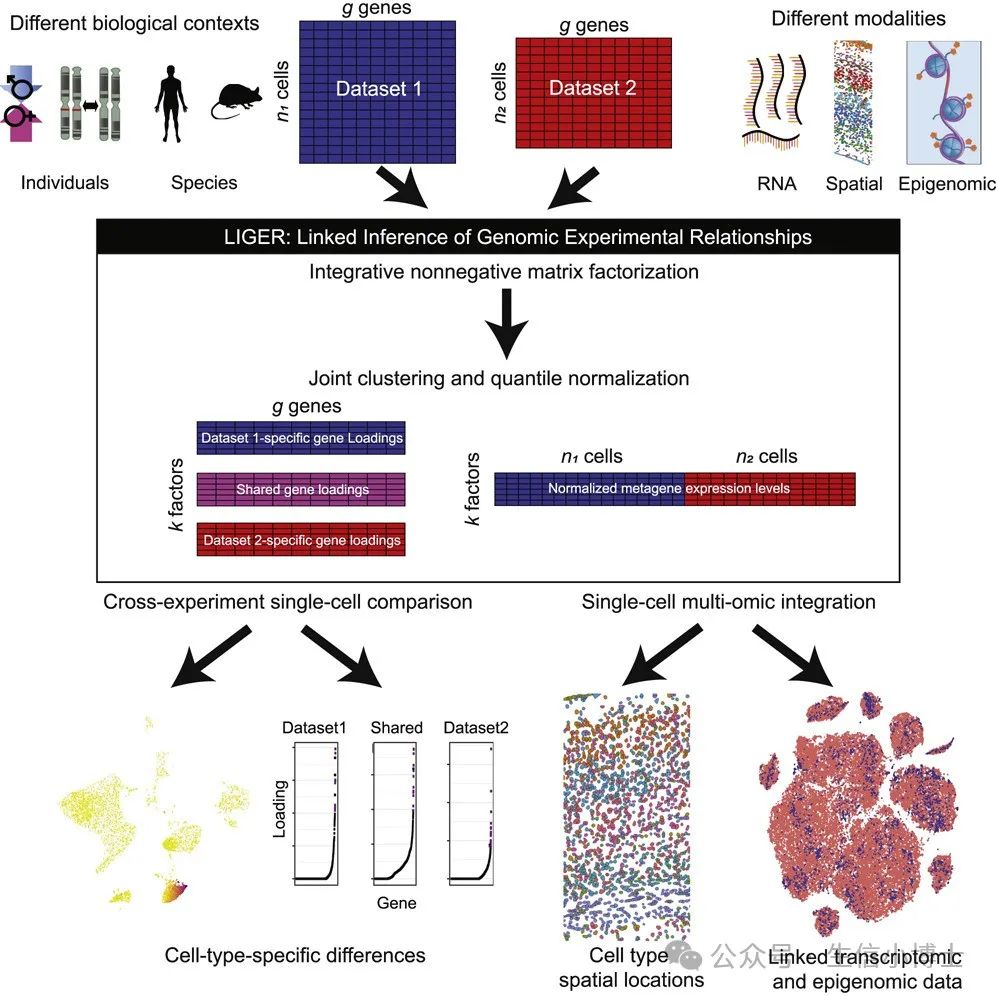
LIGER(Linked Inference of Genomic Experimental Relationships,基因实验关系的链接推断)是一个用于整合和分析多个单细胞数据集的软件包,由Macosko实验室开发,并由Welch实验室维护和扩展。它依赖于整合的非负矩阵分解技术来识别共享的和数据集特定的因子。
LIGER可以用来在多种背景下比较和对比实验数据集,例如:
-
实验批次间
-
不同个体间
-
不同性别间
-
不同组织间
-
不同物种间(例如,小鼠和人类)
-
不同模态间(例如,单细胞RNA测序和空间转录组数据,单细胞甲基化或单细胞ATAC测序)
一旦多个数据集被整合,该软件包提供了进一步的数据探索、分析和可视化的功能。用户可以:
-
识别cluster
-
找到显著的共享(和数据集特定的)基因标记
-
与之前识别的细胞类型比较簇
-
使用t-SNE和UMAP可视化簇和基因表达
让人兴奋的是,作者设计了LIGER r包,使其能够与现有的单细胞分析软件包(包括Seurat)接入。
代码实战
这里的数据仅作演示使用,读者使用自己的数据可以试一试
如果你想使用我的pbmc:
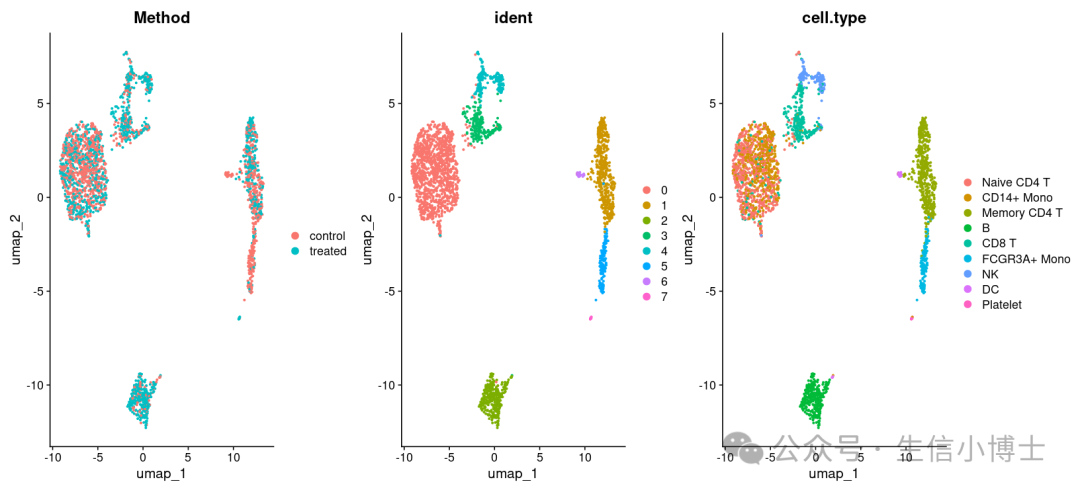
#remotes::install_github('satijalab/seurat-wrappers').libPaths(c('/home/rootyll/seurat_v5/',"/usr/local/lib/R/site-library","/usr/lib/R/site-library","/usr/lib/R/library"))#install.packages('rliger')library(rliger)library(Seurat)library(SeuratData)library(Seurat)library(SeuratWrappers)#下面正式开始-----------------------------------------------------# options(timeout = 9000)# #InstallData("pbmcsca")# data("pbmcsca")load("~/gzh/pbmc3k_final_v4.rds")pbmc$Method=pbmc$grouppbmcsca=pbmc# Please update your `liger` version to 0.5.0 or above before following this tutorialpbmcsca <- NormalizeData(pbmcsca)pbmcsca <- FindVariableFeatures(pbmcsca)pbmcsca <- ScaleData(pbmcsca, split.by = "Method", do.center = FALSE)#整合的过程就至需要下面这两句代码----pbmcsca <- RunOptimizeALS(pbmcsca, k = 20, lambda = 5, split.by = "Method")pbmcsca <- RunQuantileNorm(pbmcsca, split.by = "Method")# You can optionally perform Louvain clustering (`FindNeighbors` and `FindClusters`) after# `RunQuantileNorm` according to your needspbmcsca <- FindNeighbors(pbmcsca, reduction = "iNMF", dims = 1:20)pbmcsca <- FindClusters(pbmcsca, resolution = 0.3)# Dimensional reduction and plottingpbmcsca <- RunUMAP(pbmcsca, dims = 1:ncol(pbmcsca[["iNMF"]]), reduction = "iNMF")DimPlot(pbmcsca, group.by = c("Method", "ident", "cell.type"), ncol = 3)head(pbmc@meta.data)
LIGER还具有其他功能,后续我们再分享~
-
Iterative Single-Cell Multi-Omic Integration Using Online iNMF
-
Integrating unshared features with UINMF
-
scATAC and scRNA Integration using unshared features (UINMF)
-
Cross-species Analysis with UINMF
-
Performing Parameter Selection
-
Integrating spatial transcriptomic and transcriptomic datasets using UINMF (Click to Download)
-
Integrating Multiple Single-Cell RNA-seq Datasets
-
Jointly Defining Cell Types from scRNA-seq and scATAC-seq
-
Jointly Defining Cell Types from Single-Cell RNA-seq and DNA Methylation
-
Running Liger directly on Seurat objects using Seurat wrappers

生信小博士
【生物信息学】R语言开始,学习生信。Seurat,单细胞测序,空间转录组。 Python,scanpy,cell2location。资料分享
公众号
参考;https://github.com/welch-lab/ligerhttps://github.com/satijalab/seurat-wrappershttps://htmlpreview.github.io/?https://github.com/satijalab/seurat.wrappers/blob/master/docs/liger.html

![]()
看完记得顺手点个“在看”哦!



















 766
766

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











