







文章内容主要参考:
OrthoFinder 功能
- 查找直系同源组和直系同源物
- 推断所有直系同源组的有根基因树
- 识别基因树中的所有基因重复事件
- 推断有根物种树,并将基因复制事件从基因树映射到物种树上
- 为不同物种基因组间的比较分析提供全面的统计信息
OrthoFinder 功能及优势
- 使用 DIAMOND 软件进行 blast,大幅提高 blast 速度。
- 使用所有 orthogroups 推断物种树,而非只使用单拷贝直系同源组推断物种树,将大量的非单拷贝基因信息利用起来。
- 利用 基因复制事件的不可逆性,实现不依赖外群而生成有根物种树和有根基因树。
- 安装、使用简单且提供全面的统计信息,基本满足与进化有关相关的分析的所有需求。
- 通过修改 config.json 文件,OrthoFinder 支持用户自定义调用软件。
OrthoFinder 下载
1. 使用 conda(强烈推荐)
$ conda install orthofinder
conda 将自动安装 orthofinder 依赖的软件(如 FastTree、MAFFT 等)且调用简单(如 $ orthofinder -h)
2. github 下载
OrthoFinder 是一款用 python 编写的程序,依赖 python 和 numpy、scipy 包。若缺少上述依赖则下载 OrthoFinder.tar.gz(58.7MB),若满足则下载 OrthoFinder_source.tar.gz(4.58MB)。下载地址:https://github.com/davidemms/OrthoFinder/releases。下载完成后解压即可,通过文件夹中 orthofinder.py 调用 OrthoFinder(如 $ ./orthofinder.py -h)。
注意!若要使用 -M msa 参数,还要需要自行安装 FastTree、MAFFT 程序。
OrthoFinder 工作流程
- 使用 DIAMOND 软件 对输入序列进行 all-vs-all 序列比对。
- 使用 MCL(Markov Cluster Algorithm)算法 根据比对结果进行聚类,得到 直系同源组(orthogroup)。其中每个 orthogroup 的蛋白及序列信息存放在 Orthogroup_Sequences 文件夹中,单拷贝 orthogroup 的蛋白及序列信息存放在 Single_Copy_Orthologue_Sequences 文件夹中,orthogroup 的统计信息存放在 Comparative_Genomics_Statistics、Orthogroups 文件夹中。
如 OrthoFinder 自带案例(ExampleData)中总共包含 2733 个基因,MCL 将 2202 个基因划分为 604 个 orthogroups(gene_num > 2),剩余 531 个基因为离散点(每个基因独立成组)。 - 使用 FastMe 软件 对每个 orthogroup(gene_num >= species_num)构建 无根基因树(gene tree)。如自带案例中总共生成 324 个基因树文件。
- 使用 STAG(Species Tree Inference from All Genes)软件 根据 orthogroups(包含所有物种,如自带案例推断出的 604 个 orthogroups 中只有 316 个 orthogroups 中的同源基因在所有物种中均有分布)推断 无根物种树(species tree)。
通过参数 -M dendroblast 或 -M msa,OrthoFinder 可以调用 STAG 中两种构建物种树的方法:DendroBLAST(默认) 和 CMSA(Concatenated Multiple Sequence Alignment,联合多序列比对)。 - 使用 STRIDE(Species Tree Root Inference from Gene Duplication Events) 通过基因复制事件的不可逆性为无根物种树、无根基因树赋根,得到有根物种树、有根基因树、基因间的直系同源关系、基因复制事件。结果存放在文件夹 Species_Tree、Gene_Tree、Orthologues、Gene_Duplication_Events、Comparative_Genomics_Statistics 中。
OrthoFinder 输入文件说明
OrthoFinder 将默认读取(-f)指定文件夹内所有以 .fa、.faa、.fasta、.fas、.pep 结尾的 蛋白序列文件 作为输入。
OrthoFinder 输出文件说明
OrthoFinder 的标准输出包括:直系同源组,直系同源基因,有根基因树,解析基因树,无根物种树、有根物种树,基因重复事件以及相关的统计数据。
1. Orthogroups 文件夹
- Orthogroups.tsv、Orthogroups.txt:记录了 MCL 中 成功聚类(直系同源组中基因数 >= 2)的每个 Orthogroup 所包含的基因。
- Orthogroups_UnassignedGenes.tsv:记录了 MCL 中 未成功聚类(直系同源组中基因数 >= 1)的离散基因。
- Orthogroups.GeneCount.tsv:记录了每个 Orthogroup 中基因在物种间的分布情况,可以用于分析同源基因在物种间的收缩和扩张。
- Orthogroups_SingleCopyOrthologues.txt:记录了 单拷贝直系同源组。
2. Orthogroup_Sequences 文件夹
- 均为 FASTA 格式文件,记录了每个 Orthogroup 所包含的基因 / 蛋白的序列信息
3. Single_Copy_Orthologue_Sequences 文件夹
- 均为 FASTA 格式文件,记录了每个单拷贝 Orthogroup 所包含的基因 / 蛋白的序列信息
4. Phylogenetic_Hierarchical_Orthogroups 文件夹
- 由于复制本在进化之间存在突变速率的异质性,所以在研究同源基因时更希望所研究的同源基因来自相同的复制本。Hierarchical Orthogroups(HOG)就是为这一目的而设立的概念,HOG 指由最近共同祖先中某一基因进化而来的一组直系同源基因,进化过程中不涉及基因复制,所以 HOG 中不包含旁系同源。如下图中红框所标注的 Orthogroup。

- OrthoFinder 以物种树中的节点(LCA)为标准,寻找有根基因树内由 LCA 中基因进化来的 HOG,对原先 MCL 算法得到 orthogroup 进行细分。输出文件 N0.tsv,N1.txt,N2.tsv,… 分别指以物种树 N0,N1,N2,… 节点为标准推断出的 HOG。
5. MultipleSequenceAlignments 文件夹
- 此文件夹仅在 -M msa 模式下输出,均为 FASTA 格式文件。
- 记录了每个 orthogroup 中序列间的多序列比对结果。
- 记录了程序通过 CMSA 算法过滤后的 orthogroup 中各序列串联后的多序列比对结果,同时比对结果中空位数 > 50% 的列已被删除。
6. Species_Tree 文件夹
- SpeciesTree_rooted.txt:STAG、STRIDE 算法计算出的有根物种树结构。
- SpeciesTree_rooted_node_labels.txt:相比上树在节点处具有标签( N 0 , N 1 , . . . , N m N_0,N_1,...,N_m N0,N1,...,Nm),让后续的分析中可以方便的指定物种树节点。
- Orthogroups_for_concatenated_alignment.txt:仅在 -M msa 模式下输出,列出了所有串联起来用于推断物种树的 orthogroup ID。
7. Gene_Trees 文件夹
- 记录了每个 orthogroup(gene_num >= 4)的有根基因树结构。
8. Gene_Duplication_Events 文件夹
- 注意!OrthoFinder 只统计记录支持值(Support) >= 50% 的的复制事件。支持值是指复制后两个基因副本未被丢失的比例,Support >= 50% 表示复制后至少有一半基因在演化中保留了下来。
- Duplications.tsv:记录了程序推测出的所有基因复制事件的信息。其中 Species Tree Node 表示基因复制事件发生时所对应的物种树节点(即复制是在该物种内发生的);Gene tree node 表示基因复制事件发生时所对应的基因树节点与基因复制事件对应的节点;Support 表示复制后两个基因副本未被丢失的比例;Type 中 Terminal 表示重复发生在物种树的末端分支上,Non-Terminal 表示重复发生在物种树的内部分支上,被多个物种共享;Genes 1、Genes 2 为基因列表,其中 Genes 1 表示来自复制后基因的一个副本;Genes 2 表示来自复制后基因的另一个副本。
- SpeciesTree_Gene_Duplications_0.5_Support.txt:记录了物种树每个节点、分枝上包含的基因复制事件的总和,格式为节点或物种名 + 数字(基因复制事件数量)。
9. Orthologues 文件夹
- 以物种为单位,记录了每个物种与其他物种间的直系同源基因。
10. Comparative_Genomics_Statistics 文件夹
- Statistics_Overall.tsv:记录了有关 orthogroup 的常规统计信息。
- Statistics_PerSpecies.tsv:以物种为单位,记录了有关 orthogroup 的常规统计信息。
- Orthogroups_SpeciesOverlaps.tsv:记录了每个物种对之间共享的 orthogroup 数。
- Duplications_per_Species_Tree_Node.tsv:记录了物种树中每个节点、物种中发生基因重复事件的数量。
- Duplications_per_Orthogroup.tsv:记录了每个 orthogroup 中推断出的基因重复事件数量。
- OrthologuesStats _ *:记录了每对物种之间一对一、一对多和多对多关系的直向同源物数量。
11. WorkingDirectory 文件夹
- OrthoFinder 运行所需的必须中间文件, 如 DIAMOND 比对结果,STAG 输出的无根物种树等。
附录:
背景知识
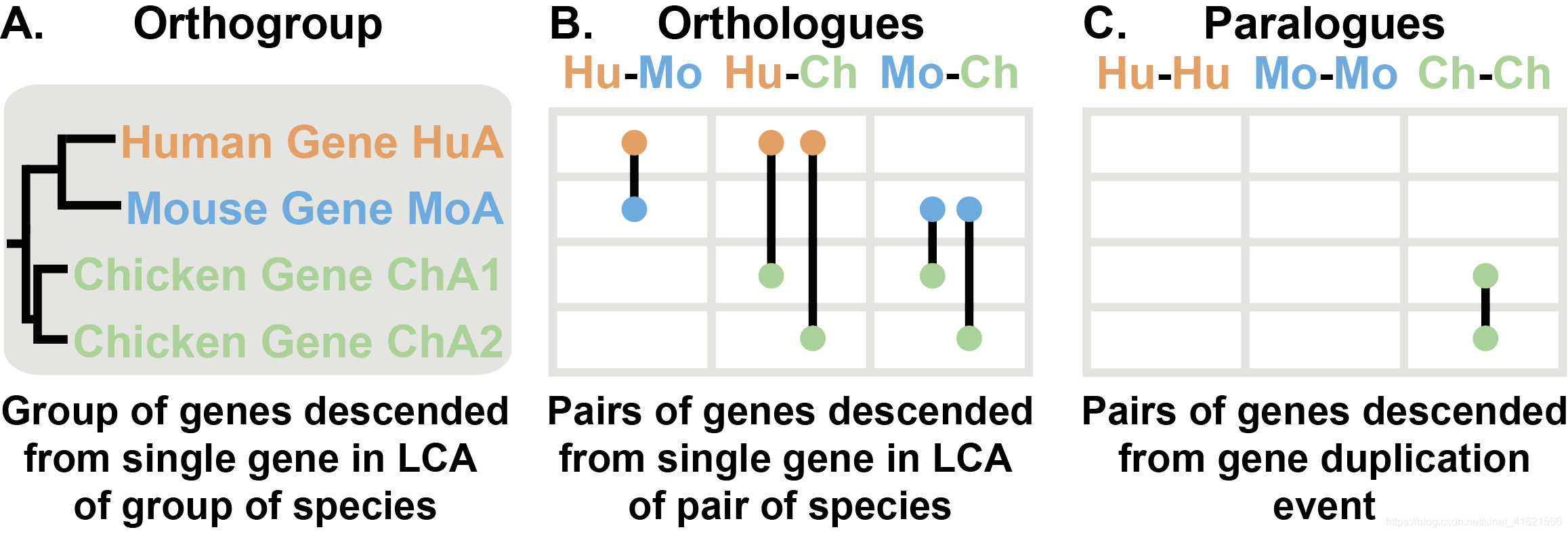
- 直系同源基因(Orthologs、Orthologues):两物种间 由最近共同祖先(last common ancestor,LCA)中某 基因进化 而得到的一组基因。如图 B 所示,在 Human 和 Mouse 间 HuA 与 MoA 是直系同源基因,在 Human 和 Chicken 间 HuA 与 ChA1、ChA2 是直系同源基因。如果有物种 Cattle 且包含 CaA1 和 CaA2 基因,则在 Cattle 和 Chicken 间 CaA1、CaA2 与 ChA1、ChA2 是直系同源基因。所以直系同源基因存在一对一、一对多、多对多三种情况。其中一对多、多对多也称为 共直系同源基因(co-orthologs)。每两对物种间的直系同源基因汇总信息存放在 OrthoFinder 输出文件夹中的 Orthologues 文件夹中。
- 直系同源组(Orthogroup):各物种间 由最近共同祖先(LCA)中某基因 进化 而得到的一组基因。如图 A 中 HuA、MoA、ChA1、ChA2 都是由同一个基因进化得到,构成直系同源组。
- 旁系同源基因(Paralogs、Paralogues):同一物种内由基因复制而产生的一组基因。如图 C 中 Chicken 内 ChA1 与 ChA2 是一对旁系同源基因。
- 基因复制(Gene Duplication):基因在物种进化过程中发生了复制。一般根据每个 orthogroup 的基因树结构,通过每次分枝后左、右枝间是否包含旁系同源基因来确定 基因复制 事件。
下图为自带案例中直系同源组 OG0000006 的有根基因树结构。首先分析 N16(node 16),其左右枝 N10、N11 是旁系同源(agal),说明 N16 发生了一次基因复制。不断递归可以发现,N19 后发生了 4 次基因复制。同理分析 N15,其中 N2、N4、N6 为旁系同源(geni),说明 N15 后发生了 2 次基因复制。结合 N15、N19,说明 N20 后发生了 6 次基因复制。由于 agal、geni 中基因与 N1 均不是旁系同源,所以 OG0000006 中总共发生了 6 次基因复制事件。
注意!OrthoFinder 只统计记录支持值(Support) >= 50% 的的复制事件。 支持值是指复制后两个基因副本未被丢失的比例,Support >= 50% 表示复制后至少有一半基因在演化中保留了下来。
- 注意!直系、旁系同源关系与基因复制事件的确定都依赖于基因树的结构,不同的基因树会导致基因间不同的直系、旁系同源关系以及不同的进化历程。直系、旁系同源关系的例子参见下图,基因复制事件的例子参见 “附录-STRIDE”。
- 讨论图中 Myc_aga 和 Myc_gen 两物种时, gi_3081 与 gi_5064、gi_5740 是直系同源关系,但与 gi_5062 则不是直系同源关系。所以只有构建好基因树,才能确定基因间的同源关系。

- 最大简约法(Maximum Parsimony):最简单的解释就是最好的解释,除非找到反例。最大简约法是系统发育分析中最常使用的一种准则,用于简化NP(Nondeterministic Polynomially,非确定性多项式)问题。
DIAMOND
DIAMOND 是 2015 年 nature methods 上发布的一款新序列比对软件,速度是 Blast 的500x-20000x,并且具有相似的准确度。
DendroBLAST
DendroBLAST 不依赖多序列比对结果 构建系统发育树,使用 -M dendroblast 调用,伪代码如下:
- 根据每个包含全部物种的 orthogroup 的 基因树,构建 物种距离矩阵 。
N
N
N 个 orthogroups 会得到
N
N
N 个物种距离矩阵。其中物种间距离是指基因树中两物种直系同源基因间的距离,如果是一对多、多对多情况则以最近基因距离作为物种距离。
如自带案例中有 316 个 orthogroups 包含全部物种,会生成 316 个物种距离矩阵,距离文件存放在 WorkingDirectory/Distances_SpeciesTree 文件夹中。 - 利用 FastMe 软件 根据物种距离矩阵推断 物种树,有 N N N 个物种距离矩阵就会推断出 N N N 个物种树。
- 将
N
N
N 个物种树整合成 1 个物种树作为最终结果。
CMSA
CMSA 是将 单拷贝直系同源基因 序列在 串联起来进行多序列比对 的输出作为推断物种树的输入,使用 -M msa 调用。由于基因复制和丢失事件经常发生,所以 单拷贝直系同源组 是稀少的,尤其当分析大量物种时。作者对 CMSA 算法进行了优化:OrthoFinder 用单拷贝物种数超过总物种数 p% 的 orthogroup 推断物种树。如总物种数是 50,p% = 60%,所有 orthogroups 中单拷贝物种数超过 30 的 orthogroup 都将被用于推断物种树。伪代码如下:
- 设总物种数为 S S S,单拷贝直系同源组数为 n 1 n_1 n1,若 n 1 > = 1000 n_1>=1000 n1>=1000 程序终止并使用这 n 1 n_1 n1 个直系同源组推断物种树,若 n 1 < 1000 n_1<1000 n1<1000 进行下面步骤。
- 令 s 1 = S s_1=S s1=S 开始循环: s 2 = s 1 − 1 s_2=s_1-1 s2=s1−1,单拷贝物种数 > = s 2 >=s_2 >=s2 的 orthogroups 数目 n 2 n_2 n2,计算 n 2 / n 1 – 1 n_2/n_1 – 1 n2/n1–1, 1 − s 2 / s 1 1 - s_2/s_1 1−s2/s1;令 n 1 = n 2 n_1=n_2 n1=n2;若 n 2 > = 100 n_2 >=100 n2>=100 且 n 2 / n 1 – 1 < 2 ( 1 – s 2 / s 1 ) n_2/n_1 – 1 < 2(1 – s_2/s_1) n2/n1–1<2(1–s2/s1),循环终止并使用这 n 2 n_2 n2 个 orthogroups 推断物种树。作者以 2 ( 1 – s 2 / s 1 ) > n 2 / n 1 – 1 2(1 – s_2/s_1) > n_2/n_1 – 1 2(1–s2/s1)>n2/n1–1 作为阈值保证筛选出来的 orthogroup 内有足够数量的单拷贝直系同源基因。循环终止时 s 2 / S = p % s_2/S=p\% s2/S=p%。
- 将步骤 2 中循环终止时筛选出的 n 2 n_2 n2 个 orthogroups 中的 单拷贝基因 序列以物种为单位串联起来并使用 MAFFT 进行 多序列比对,结果存放在文件夹 MultipleSequenceAlignments 中。如 S = 30 S=30 S=30, n 2 = 50 n_2=50 n2=50 ,将50组 orthogroups 中的单拷贝基因的序列串联起来,得到 30 个序列,每条序列既代表一个物种。使用 MAFFT 对这 30 个序列进行多序列比对。
- 步骤 2 中筛选出来的 orthogroups 中,有旁系同源的基因不会被串联进物种序列。如 FigA 中 ChA1、ChA2 不会被串联进 Chicken 的物种序列上,而 HuA、MoA 则会分别被串联进 Human、Mouse 的物种序列中。所以步骤 3 的多序列比对结果中会出现大量的空位(gap)。作者将空位数超过
S
−
0.5
s
2
S-0.5s_2
S−0.5s2 的列删去,以剩下的多序列比对结果作为 FastTree 的输入来推断 物种树。列筛选步骤不仅进一步筛选了 orthogroups,同时还删去了多序列比对结果中较差的区间。
STRIDE
参考文献:STRIDE: Species Tree Root Inference from Gene Duplication Events
STRIDE 利用 基因复制事件的不可逆性,推断基因树、物种树的生根位点。程序以无根物种树、无根基因树作为输入,具体算法如下:
- 遍历无根物种树的边,得到所有可能的有根物种树结构。如下图中 A 为无根物种树结构,B-F 为所有可能的有根物种树结构。

- 将无根基因树按照有根物种树的结构绘制成有根基因树。如下图 A 为无根基因树结构,B-F 为根据物种树 B-F 绘制的有根基因树。图中 D D D 表示基因复制事件, L L L 表示基因丢失事件。图 G、H 分别为根据基因树 C、D 推断出的基因间直系同源关系表,可以发现不同结构的基因树会推断出不同的结果。所以 有根物种树决定了有根基因树的结构与基因间的直系、旁系同源关系 。

- 以基因树 C 为例说明基因复制、丢失事件的判断方法。首先因为基因复制分成了黄紫、绿两部分。再分析黄紫枝,如果黄紫基因由共同祖先中某基因进化得到,其局部的基因树结构应该与物种树 C 一致,由于结构不一致,说明黄紫是分别由共同祖先内两个旁系同源基因分别进化得到,即发生了基因复制。所以在基因树 C 中总共发生了 2 次基因复制事件,并且都发生在共同祖先内,即每个物种在未发生基因丢失的情况下应该有 3 个旁系同源基因。由于象、狼只有 2 个旁系同源基因,同时象、狼的共同祖先即为 4 个物种的共同祖先,所以基因丢失事件只能发生在象、狼的进化过程中,即发生了 2 次基因丢失事件。由于鱼、鸟中是单拷贝,同时他们的共同祖先并非 4 个物种的共同祖先,所以基因丢失事件既可能发生在鱼、鸟的进化过程中,也可能发生在共同祖先的进化过程中。如果发生在鱼、鸟共同祖先的进化过程中则发生了 2 次基因丢失事件,如果发生在鱼、鸟进化过程中则发生了 4 次基因丢失事件。根据最大简约法推断出基因丢失发生在鱼、鸟共同祖先的进化过程中。综上,基因树 C 中总共发生了 2 次基因复制,4 次基因丢失。图中标注了事件发生的时间。
- 根据每个有根基因树结构(基因树 B-F)中基因复制、基因丢失事件发生的次数,计算每种结构出现的概率。遍历所有的 orthogroup,如果有 N 个 orthogroup 则每个有根物种树(物种树 B-F)会计算出 N 个概率。如物种树 B 有概率集 { p b 1 , p b 2 , p b 3 , . . . , p b N } \{p_{\ b_1},p_{\ b_2},p_{\ b_3},...,p_{\ b_N}\} {p b1,p b2,p b3,...,p bN} 。整合这 N 个概率(公式参见文献),得到物种树的出现概率,即在此边生根的概率。
- 计算所有有根物种树的出现概率,即所有边的生根概率(如下图)。以概率最大的边生根得到的物种树作为有根物种树的输出,以该物种树下得到的各 orthogroup 基因树作为有根基因树的输出。同时,统计并输出基因复制、orthogroup 基因间直系同源关系等信息 。

为了方便理解,上述描述的算法与 STRIDE 实际使用的算法有一定出入,详细请参见文献。

















 1605
1605

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









