










 本文概述了非编码RNA的分类,重点介绍基于RNA二级结构和机器学习(如SVM)的鉴定策略。通过选择合理的初始特征集合,如ORF覆盖率和同源性特征,以及特征选择的过程,展示了如何使用SVM进行编码/非编码RNA的区分。
本文概述了非编码RNA的分类,重点介绍基于RNA二级结构和机器学习(如SVM)的鉴定策略。通过选择合理的初始特征集合,如ORF覆盖率和同源性特征,以及特征选择的过程,展示了如何使用SVM进行编码/非编码RNA的区分。
第六章 非编码RNA鉴定
主要为RNA-seq相关知识,部分内容作笔记自查使用。如有错误或遗漏还请海涵,可评论或邮箱联系。
最后修改时间:2020-09-07 14:38:07 星期一
非编码RNA分类
非编码RNA,是指不需要翻译为蛋白,在RNA形式下即可行使其生物学功能的RNA分子。非编码RNA通常彼此协同作用,共同调控细胞生长、发育、凋亡等一系列重要的生理过程。
- 负责维系细胞基础代谢(housekeeping genes)
- rRNA
- tRNA
- ...
- 调控其他基因的转录和翻译
- miRNA
成熟miRNA通过碱基互补配对,识别特定的目标RNA,下调其表达,从而达到对特定生物学过程进行调控的效果。miRNA在多种肿瘤的发生、发展中起核心调控作用,可以作为病程诊断与进展程度的marker - lncRNA(long non-coding RNA)
长度达几十乃至上百bp,包含多个exon,有可变剪切和polyA尾巴
- miRNA
目前的研究重点是,对新找到的lncRNA的鉴定和功能分析
非编码RNA鉴定
-
基于RNA二级结构来鉴定新的非编码RNA
优点:速度快。pre-miRNA会形成发卡结构,tRNA是三脚结构,均可以通过二级结构确定
缺点:lncRNA的功能不依赖于特定二级结构,不适用 -
使用SVM等机器学习方法分类
不依赖多序列比对等外部信息,只使用转录本序列本身的信息,对miRNA和lncRNA同样适用,速度较快。
使用SVM分类步骤
一、 确定初始特征集合(feature set)
选择一个合理而有效的初始特征集合,对后续的feature selection乃至identification的效果都非常重要。若初始集合中混入不相关的feature,会严重影响后续feature selection的效率。
实践中主要会依据已有的文献、数据,结合自己的生物学直觉进行。以Coding Potential Calculator为例。根据相关文献并结合生物学背景,选取60个RNA序列水平的特征,作为初始特征集。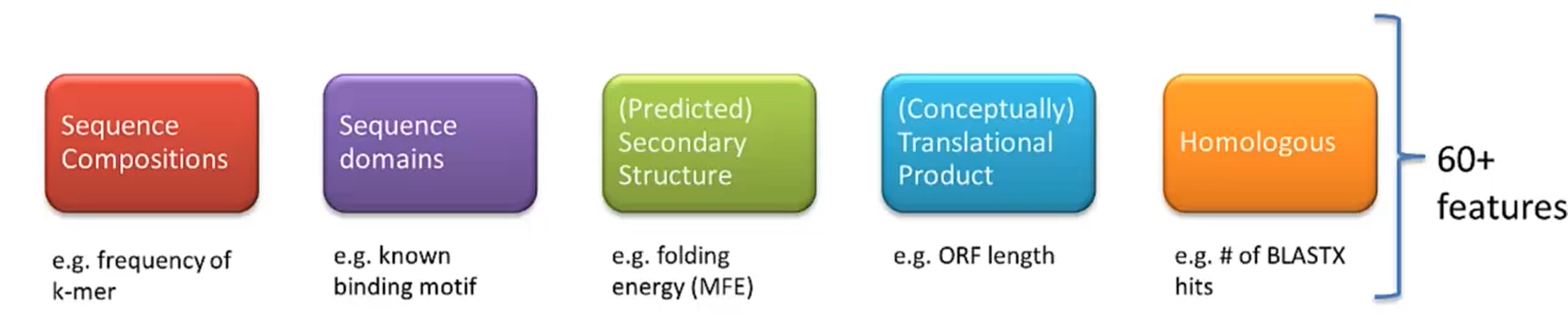
二、 特征选择(feature selection)
序列特征有很多种类可供选择,如下图所示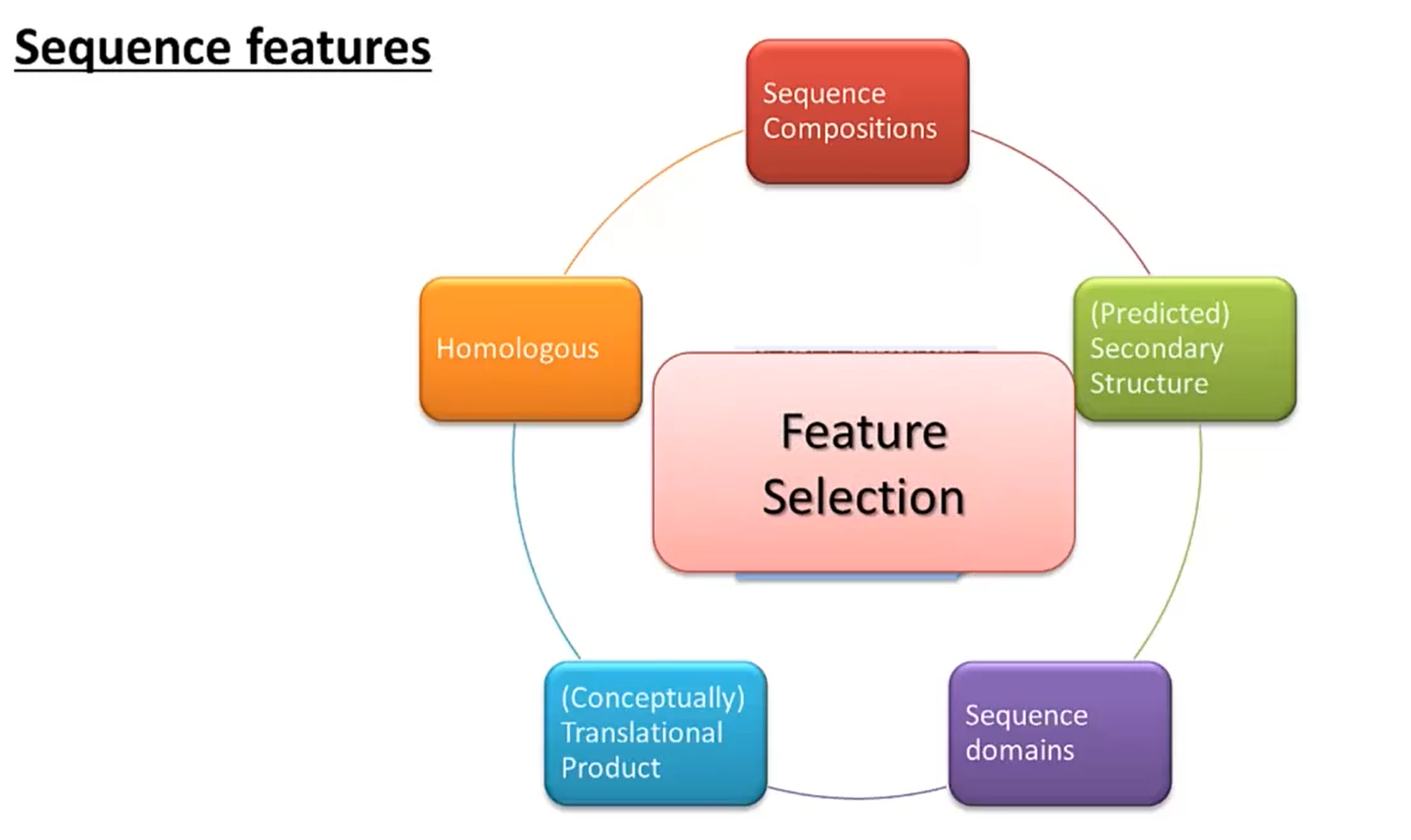
我们需要对如此多的特征进行筛选,得到针对特定分类目的表现较好的特征子集。我们希望在确保准确性的前提下,这个子集尽可能小,以加快计算速度。
紧接步骤1。首先利用前向搜索算法,从中筛选出11个feature作为初步特征子集。此时集合元素较少,可以基于广度优先策略进行完全搜索,最终得到6个features
其中,蓝色的3个特征,是基于RNA序列中利用概率模型预测得到的ORF的
- Coverage
- 预测得到的ORF占整个RNA序列长度的比例
- ORF Integrity
- 预测的ORF是否是完整的
- LOG-ODD score
- 对预测可靠性的评估。分数越高,预测得到的ORF越可靠
黄色的3个特征是基于同源性信息的
基本的想法是,编码蛋白的mRNA,较不编码的non-coding RNA,会有更大的可能在蛋白数据库搜索中找到相似的蛋白。
即使non-coding RNA随机地匹配上若干蛋白片段,但因为其中没有真实的ORF,因此匹配会随机分散在多个区域,而不是一个特定的区域。
2.1 特征选择的方法
- 完全搜索
- 广度优先搜索
对原始特征集合中所有可能的组合进行穷尽测试。但考虑到组合爆炸问题,实际应用中并不适用于很大的初始feature set。
- 广度优先搜索
- 启发式搜索
- 前向搜索(SFS)
贪心思想。向空子集中依次尝试加入单个feature,留下表现最好的那个。接着再依次尝试加入剩余的单个feature,直到新加入的feature无法继续提升分类准确度为止。
由于前向搜索中不能删除已经被选择的feature,可能会导致高度相关的feature同时被加入,造成冗余。 - 后向搜索(SBS)
贪心思想。向全集中依次尝试删除单个feature,留下表现最好的子集。接着再依次尝试删除剩余的单个feature,直到新删除的feature无法继续提升分类准确度为止。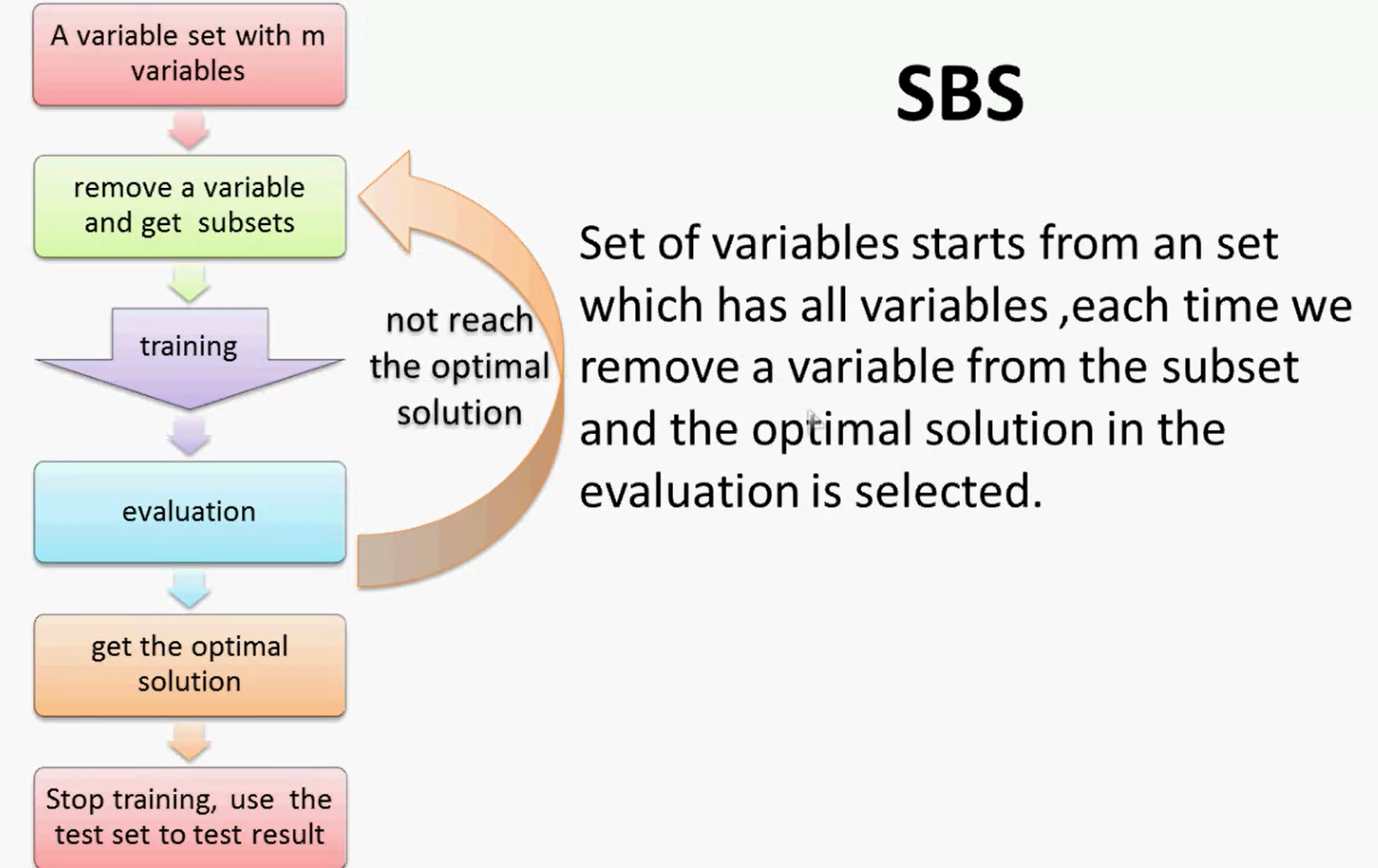
- 增L去R(LRS)
每次训练时,增加L个特征,同时删除R个特征,L和R固定。该方法可以避免前/后向搜索中的信息重复
- 前向搜索(SFS)
- 随机搜索
- 模拟退火算法
引入随机因素以避免陷入局部最优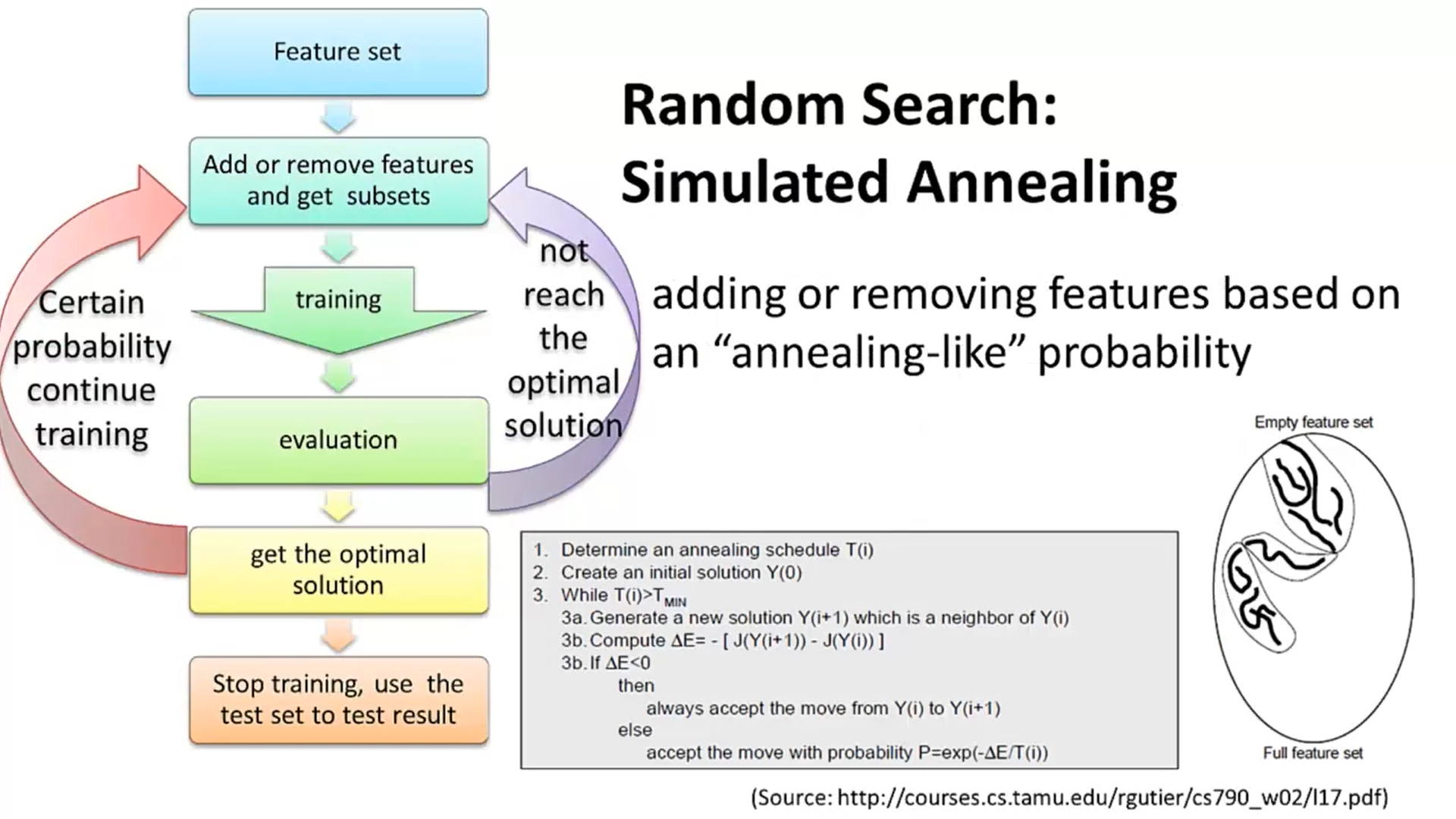
- 序列浮动选择
从增L去R算法改进而来,每次增加的L和删除的R不是一个定值,而是浮动的。
- 模拟退火算法
三、 训练SVM
将6个feature组成的训练集输入到SVM模型中进行训练,得到能做编码/非编码RNA二分类的机器学习模型


















 42
42

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











