







1. 什么是单倍型?
image
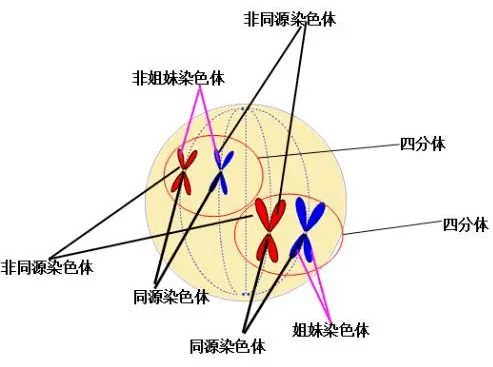
同源染色体:同源染色体,一个来自母本,一个来自于父本。
单倍型:单倍体基因型的简称。遗传学上指在单条染色体上一系列遗传变异位点的组合。
2. 单倍型组装的意义?
目前,大多数二倍体基因组组装都忽略了同源染色体之间的差异,将基因组组装成一个假的单倍体序列,这是二倍体类型的组装的人为共识。这种人为的共识可能导致基因注释的不精确和生物学解释的错误。
为了深入研究的需要,更多的物种需要将来自父母的遗传信息都获得,因此参考基因组就需要获得两个单倍体基因组,也就是单倍型基因组。
目前单倍型技术主要应用领域包括:
- 在医学上探索致病机理,挖掘致病基因,寻找疾病治疗新方法;
- 在群体遗传学上分析等位基因间差异,追踪个体亲缘关系,了解生物迁徙模式和进化历史;
- 在农业上发掘优异等位基因变异,探索杂种优势理论等。
3. 如何进行单倍型组装?
早期已经提出了几种算法来生成
 最低0.47元/天 解锁文章
最低0.47元/天 解锁文章



















 2354
2354

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











