







分析目标:
(1)梳理WGCNA的基本流程。
(2)功能注释
(3)对相应的基因模块进行时空表达特征评估
一、WGCNA分析(基因共表达分析)
我们有4000+个感兴趣的基因,希望通过这一步得到的结果是:按照基因之间的表达特征的相似性,将其分为若干基因模块(module)。
本次实验使用的数据集
(1)GSE25219-GPL5175数据集:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE25219
(2)GSE25219-GPL5175样本注释:根据matrix前面的注释行,对样本数据进行提取后的结果。
注:GPL5175是一种transcript水平的检测平台,因此每一个行的标签代表的是一个基因的转录本的类型*。
(一)输入数据集的预处理
- 原始数据集
我们下载到原始数据之后,需要对探针名进行转换。GPL5175平台比较复杂的一个点在于,其注释数据中对应基因混乱不明确,给我们下游的分析带来了困难。
data<-read.table("GSE25219-GPL5175_series_matrix.txt",comment.char = "!",header = T,row.names = 1)
# ## transform the gene probe##
gene_anno<-read.csv("probe_anno.csv",row.names = 1) #transcript or gene level is important
colnames(gene_anno)<-c("ID","anno")
# ## 将row name赋值给data #方便下游的合并去交集
data2<-cbind(rownames(data),data)
colnames(data2)[1]<-"ID"
dim(data2) #17565 #1341
# ## make the ID to the rownames of this data frame
merge_data<-merge(data2,gene_anno) #将两者merge到一起
merge_data2<-merge_data[!duplicated(merge_data$anno),] #去除重复
rownames(merge_data2)<-trimws(merge_data2$anno, which = c("both", "left", "right"), whitespace = "[ \t\r\n]") #去除前后可能的空白
merge_data3<-merge_data2[,c(-1,-1342)]
dim(merge_data3) #17337 #deplicate is not too much
write.table(merge_data3,"merge_data.txt")
得到上述探针名进行基因名的转换后的数据集后,下次只需要读取转换后的矩阵即可。
data<-read.table("merge_data.txt")

- 样本数据集
这个是根据原始数据集matrix前对样本注释信息提取到的样本的注释集。
## construct the datTraits
label<-read.table("label.txt")
region<-read.table("region.txt")
year<-read.table("year.txt")
stage<-read.table("stage.txt")
meta.data<-rbind(label,region,year,stage)
meta.data2<-as.data.farme(t(meta.data))
colnames(meta.data2)<-c("ID_REF","subRegion","time")
NCX<-c("V1C","A1C","DFC"……)#等等
meta.data2[meta.data2%in%NCX,"Region"]<-"NCX"
write.csv(meta.data2,"meta.data.csv")
同样,将样本信息进行上述处理,保存到meta.data的文件中之后,我们下次如果使用只需要读取即可。
meta.data<-read.csv("meta.data.csv")
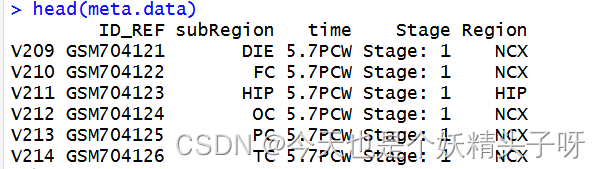
(二)对数据集进行筛选
- 基因层面的筛选
虽然原始的数据集中有很多的基因,但是我们实际上感兴趣的是这4000多个基因,因此我们需要首先,从原始数据集中提取这4000多个基因的表达情况。
genes<-read.table("input_interest_gene_all.txt",sep="\t",header = F)
genes2<-genes[!duplicated(genes$V1),]
filter_data<-data[rownames(data)%in%genes2,] #筛选感兴趣的gene set
- 样本层面的筛选
剔除属于Stage1和Stage2阶段的样本
filter_sample<-meta.data[which(!meta.data$Stage%in%c("Stage: 1","Stage: 2")),"ID_REF"]
filter_data<-filter_data[,colnames(filter_data)%in%filter_sample]
filter_metadata<-meta.data[meta.data$ID_REF%in%filter_sample,]
最后得到在行和列层面上筛选过的data和meta.data。
(三)WGCNA分析——寻找共表达的基因模块
一般网上对于这个的描述特别的杂乱和复杂,在我看来这一步可以简化为以下。
(1)加载WGCNA包
library(WGCNA)
options(stringsAsFactors = FALSE)
(2)准备基因的输入矩阵
datExpr0<-t(datExpr)
(3)筛选软阈值
## pick softscores
powers = c(c(1:20))
sft = pickSoftThreshold(datExpr1,powerVector = powers, verbose = 5)#verbose=5说明什么
#(1)是否符合幂律分布;
plot(sft$fitIndices[,1], -sign(sft$fitIndices[,3])*sft$fitIndices[,2],
xlab="Soft Threshold (power)",ylab="Scale Free Topology Model Fit,signed R^2",type="n",main = paste("Scale independence"))
text(sft$fitIndices[,1], -sign(sft$fitIndices[,3])*sft$fitIndices[,2],
labels=powers,cex=cex1,col="red")
abline(h=0.90,col="red")
#(2)节点的平均连接度
plot(sft$fitIndices[,1], sft$fitIndices[,5],
xlab="Soft Threshold (power)",ylab="Mean Connectivity", type="n",
main = paste("Mean connectivity"))
text(sft$fitIndices[,1], sft$fitIndices[,5], labels=powers, cex=cex1,col="red")
par(mfrow = c(1,1))
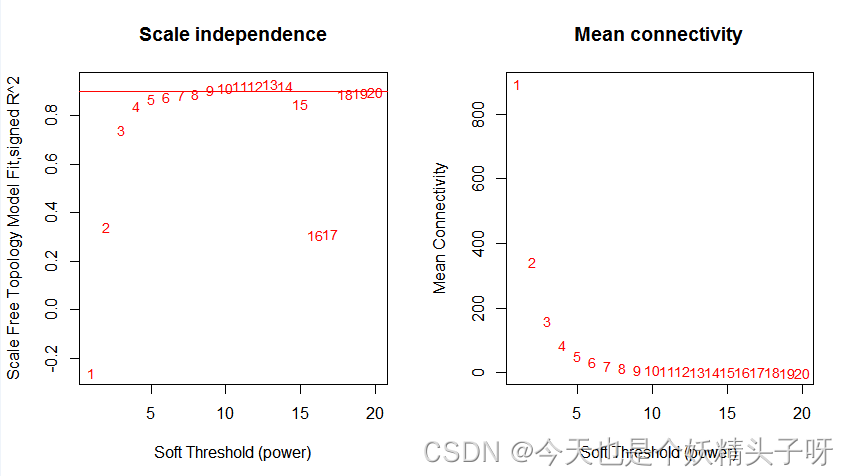 通过上述实验,筛选出软阈值为9。以9为阈值,用于下游的分析。
通过上述实验,筛选出软阈值为9。以9为阈值,用于下游的分析。
(4)一步式计算基因模块
net = blockwiseModules(datExpr1, power = 9,
corType = "pearson",
networkType="unsigned",
TOMType = "unsigned",
minModuleSize = 30,
verbose = 3) #记住power依旧是9 #这里的参数实际上是由上面一步步得到的
这一步就得到了wgcna分析的所有结果,下面可以根据这个net变量的结果进行多样化的绘图。
当需要将基因模块与表型建立起相关性时,还需要构建表型矩阵,由于这里这一部分并非是我们的核心,因此暂时不去考虑。
如对基因模块从表达特征上进行聚类。
MEDiss = 1-cor(net$MEs)
METree = hclust(as.dist(MEDiss), method = "average")
sizeGrWindow(7, 6)
plot(METree, main = "Clustering of module eigengenes",
xlab = "", sub = "")
abline(h=0.25, col = "red") #根据实际情况而定

(5)提取每一种颜色模块中的基因。
moduleGenes = names(net$colors)[net$colors%in%c("turquoise")]
write.csv(moduleGenes,"turquoise_module.csv")
二、对模块基因进行GO功能注释
(略)
三、基因模块表达的时空pattern的分析
(一)从WGCNA的结果中提取特定模块的eigengenes
这一步是对前面步骤结果的提取。
moduleColors<-as.data.frame(table(net$colors))$Var1
MEs0 = moduleEigengenes(net$MEs, moduleColors)$eigengenes
(二)与样本注释数据集合并(以提取黄色模块为例)
pcaPlotDat <- merge(filter_metadata,cbind(PC1=filter_data$MEyellow,ID_REF=rownames(filter_data)))
pcaPlotDat$PC1<-as.numeric(pcaPlotDat$PC1)
subtypeOrder<-c("Stage: 3","Stage: 4","Stage: 5","Stage: 6","Stage: 7",
"Stage: 8","Stage: 9","Stage: 10","Stage: 11","Stage: 12","Stage: 13","Stage: 14","Stage: 15")
pcaPlotDat$Stage<-factor(pcaPlotDat$Stage,levels = subtypeOrder)
(三)画图
library(ggplot2)
ggplot(pcaPlotDat,aes(x=Stage,y=PC1,group=Region))+
geom_point(size=3,aes(colour=Region,shape=Region))+
geom_smooth(method="loess",se=FALSE,aes(colour=Region),formula = y~x)+
labs(title = "Yellow module")+
theme(plot.title=element_text(hjust=0.5))
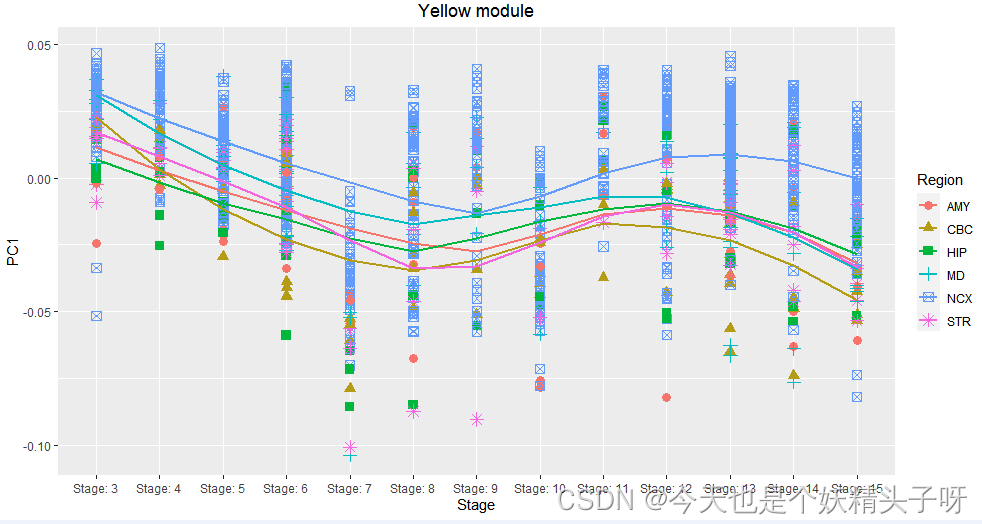 觉得得到的结果比较奇怪,和老师的plot相比。我不理解,为什么我的这个PC1有正有负,且和之前看到的与表型相关性的结果,看起来不太一样(很不明显)。
觉得得到的结果比较奇怪,和老师的plot相比。我不理解,为什么我的这个PC1有正有负,且和之前看到的与表型相关性的结果,看起来不太一样(很不明显)。
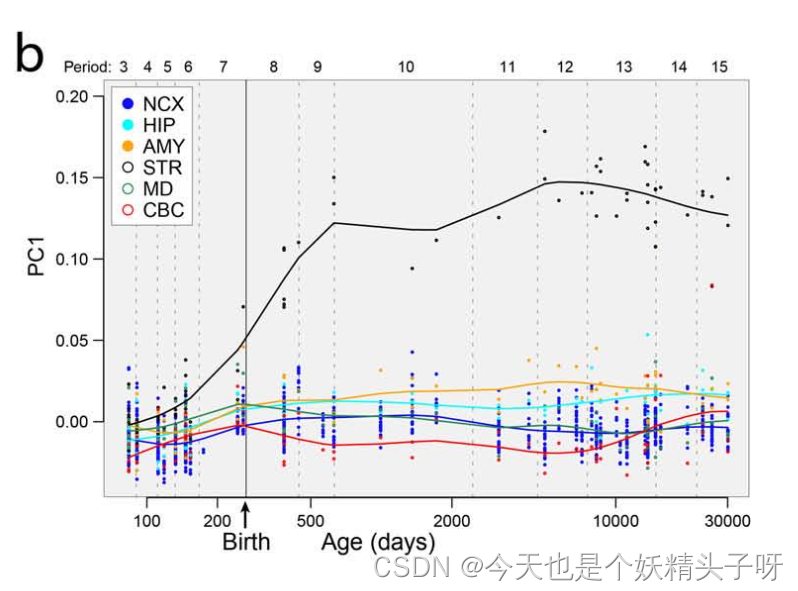 我觉得基本思路应该没问题了,可能还是一些细节的问题。
我觉得基本思路应该没问题了,可能还是一些细节的问题。
等下换一个环境,重新做一遍。

















 1万+
1万+

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









