








关注公众号,桓峰基因
这期继续补充一下转录组高级分析内容之一的筛选Hub基因,这个模块在 SCI 文章中也是经常出现,并且很多文章也是直接作为文章的两点来分析的,现在就介绍一下这部分的内容该怎么分析?
前言
我们在分析 WGCNA 之后获得了几个基因模块,但是发现基因仍然很多,需要进一步筛选基因与表型相关的基因,那么今天就介绍一下 Hub 基因,那么什么是 Hub Genes呢?在这样的网络中,高度连接的基因被称为枢纽基因,有望在理解应激/条件下反应的生物学机制中发挥重要作用。信息基因的选择是基因表达研究中的一个重要问题。由于样本量小,基因表达数据中的基因数量多,使得选择过程复杂。此外,所选的信息基因可能作为基因共表达网络分析的重要输入。此外,基因共表达网络中枢纽基因和模块相互作用的识别还有待进一步研究。本文提出了一种基于支持向量机算法从高维基因表达数据中选择信息丰富的基因的方法。此外,还试图开发一种统计方法来识别基因共表达网络中的枢纽基因。此外,在病例与对照研究中,还提出了一种基于基因连接性的差异枢纽基因分析方法。

实例解读
hub基因,是degree高的gene,在基因表达网络中有高的连接度degree,不涉及betweeness等。并且hub基因的筛选有很大的人为因素,到底是取前5%还是10%没有具体要求,一般建议5%。也就是说这是一个很宽松的设定。
基于这个建议的方法,一个R包,即dhga已经开发出来了。在三种不同的作物微阵列数据集上,比较了所提出的基因选择技术和hub基因识别方法的性能。在选择稳健信息基因集方面,所提出的基因选择技术优于现有的大多数技术。与现有的hub基因识别方法相比,该方法识别出的hub基因数量较少,符合真实网络的无标度原则。本研究报道了拟南芥的一些关键基因及其同源基因,可用于大豆铝毒胁迫响应工程。通过对多个关键基因的功能分析,揭示了大豆铝毒胁迫响应的分子机制。
https://cran.r-project.org/web/packages/dhga
1. 数据读取
我们的例子使用的是结直肠癌与正常组织差异分析获得的结果,先看下数据情况,然后将肿瘤样本和正常样本分开,如下:
Group <- read.table("DEG-group.xls", header = T, check.names = F)
NT <- Group[Group$Group %in% "NT", ]$Sample
TP <- Group[Group$Group %in% "TP", ]$Sample
DEG <- read.table("DEG-resdata.xls", header = T, check.names = F, row.names = 1)
exp <- DEG[, 7:ncol(DEG)]
NTmatrix <- exp[, colnames(exp) %in% NT]
TPmatrix <- exp[, colnames(exp) %in% TP]
head(NTmatrix[, 1:2])
## TCGA-A6-2671-11A-01R-A32Z-07 TCGA-A6-2675-11A-01R-1723-07
## ENSG00000142959 8177 2165
## ENSG00000163815 2863 3403
## ENSG00000107611 29 67
## ENSG00000162461 862 155
## ENSG00000163959 882 473
## ENSG00000144410 3 10
2. 软件安装
if (!require(dhga)) {
install.packages("dhga")
}
if (!require(WGCNA)) {
BiocManager::install("WGCNA")
}
library(WGCNA)
library(dhga)
3. Hub 基因筛选
1. WeightedGeneScore{dhga}
该函数使用软阈值参数计算邻接矩阵来构建一个基因共表达网络
beta = 6
threshold = 0.4
adj <- Adjacency(NTmatrix, 6, 0.7)
head(adj$EdgeList)
## Node_i Node_j Source
## 1 ENSG00000107611 ENSG00000144410 Predicted Interaction
## 2 ENSG00000107611 ENSG00000257335 Predicted Interaction
## 3 ENSG00000144410 ENSG00000257335 Predicted Interaction
## 4 ENSG00000107611 ENSG00000118137 Predicted Interaction
## 5 ENSG00000144410 ENSG00000118137 Predicted Interaction
## 6 ENSG00000257335 ENSG00000118137 Predicted Interaction
基因共表达网络中基因的差异Hub状态
diffhub <- DiffHub(TPmatrix, NTmatrix, 18, 18, 80, 6, alpha = 1e-04, plot = TRUE)
head(diffhub)
## Genes pval_stres pval_control
## 1 ENSG00000142959 0.570427490350264 5.86237432061609e-14
## 2 ENSG00000163815 7.54955354865796e-14 1.71220007840703e-07
## 3 ENSG00000107611 8.97877318402061e-15 5.86237432061609e-14
## 4 ENSG00000162461 8.4775260388662e-07 1.81877856225649e-14
## 5 ENSG00000163959 4.81763836844215e-08 1.62783799349465e-14
## 6 ENSG00000144410 6.66875387120388e-07 1.91298989956118e-13
## HubStatus
## 1 Unique Hub to Normal
## 2 Housekeeping Hub
## 3 Housekeeping Hub
## 4 Housekeeping Hub
## 5 Housekeeping Hub
## 6 Housekeeping Hub
基于基因连接显著性值的基因共表达网络中Hub基因的识别
x = as.data.frame(TPmatrix)
beta = 6
m = 18
s = 80
n = 20
hub.pval <- hub.pval.cutoff(x, beta, m, s, n)
基于加权基因得分的基因共表达网络中Hub基因的识别,如下:
hub.wgs(TPmatrix, beta = 6, n = 20)
## [1] "ENSG00000279544" "ENSG00000257279" "ENSG00000233783" "ENSG00000269821"
## [5] "ENSG00000228663" "ENSG00000264546" "ENSG00000235522" "ENSG00000214188"
## [9] "ENSG00000280392" "ENSG00000274554" "ENSG00000188451" "ENSG00000243053"
## [13] "ENSG00000223528" "ENSG00000280069" "ENSG00000183562" "ENSG00000272219"
## [17] "ENSG00000260034" "ENSG00000260558" "ENSG00000248483" "ENSG00000280269"
## attr(,"class")
## [1] "Hub Genes"
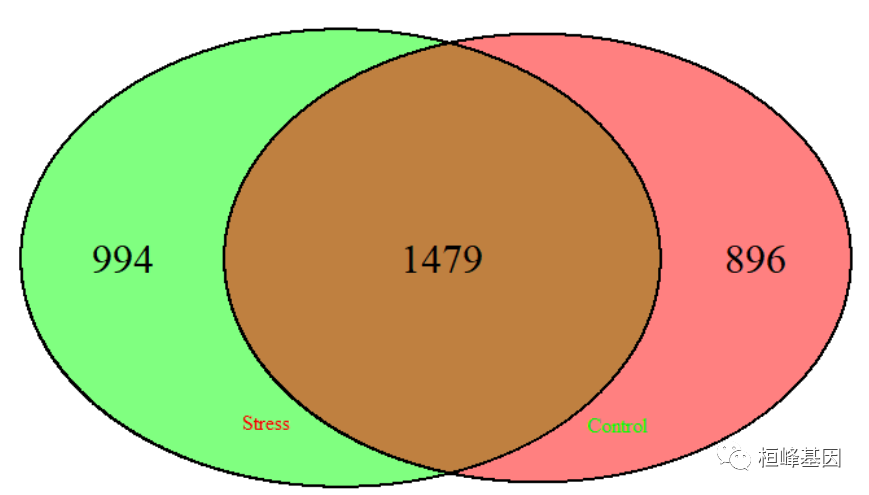
基因共表达网络中基因连接显著值的计算(pvalue.hub),然后绘制Venn图,如下:
pval.stres <- pvalue.hub(TPmatrix, beta = 6, m = 18, s = 80, plot = FALSE)
pvalue.stress <- pval.stres[, 2]
pval.control <- pvalue.hub(NTmatrix, beta = 6, m = 18, s = 80, plot = FALSE)
pvalue.control <- pval.control[, 2]
HubPlot(pvalue.stress, pvalue.control, alpha = 1e-04)
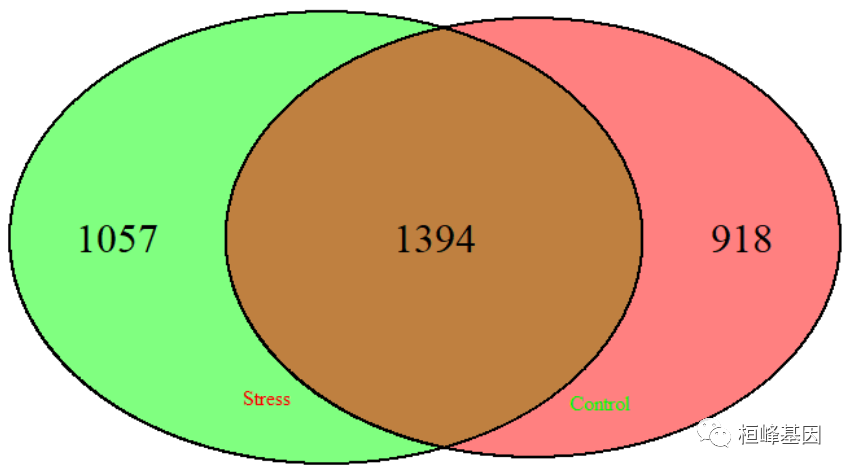
基因在基因共表达网络中的Hub状态,如下:
pval.stres <- pvalue.hub(TPmatrix, beta = 6, m = 18, s = 80, plot = FALSE)
p1 <- pval.stres[, 2]
pval.control <- pvalue.hub(NTmatrix, beta = 6, m = 18, s = 80, plot = FALSE)
p2 <- pval.control[, 2]
hs <- HubStatus(p1, p2, alpha = 1e-04)
head(hs$hub.status)
## $hub.status
## Genes Hub Status
## 1 ENSG00000142959 Unique Hub to Normal
## 2 ENSG00000163815 Housekeeping Hub
## 3 ENSG00000107611 Housekeeping Hub
## 4 ENSG00000162461 Unique Hub to Normal
## 5 ENSG00000163959 Housekeeping Hub
## 6 ENSG00000144410 Unique Hub to Normal
## 7 ENSG00000118777 Unique Hub to Normal
## 8 ENSG00000040199 Housekeeping Hub
## 9 ENSG00000120498 Unique Hub to Normal
## 10 ENSG00000036672 Unique Hub to Normal
## 11 ENSG00000169764 Housekeeping Hub
## $out1
##
## Housekeeping Hub Not Hub Unique Hub to Normal
## 1446 757 1086
## Unique Hub to Stress
## 839
基因共表达网络中基因加权得分的计算(WeightedGeneScore)
wgs <- WeightedGeneScore(NTmatrix, beta, plot = TRUE)
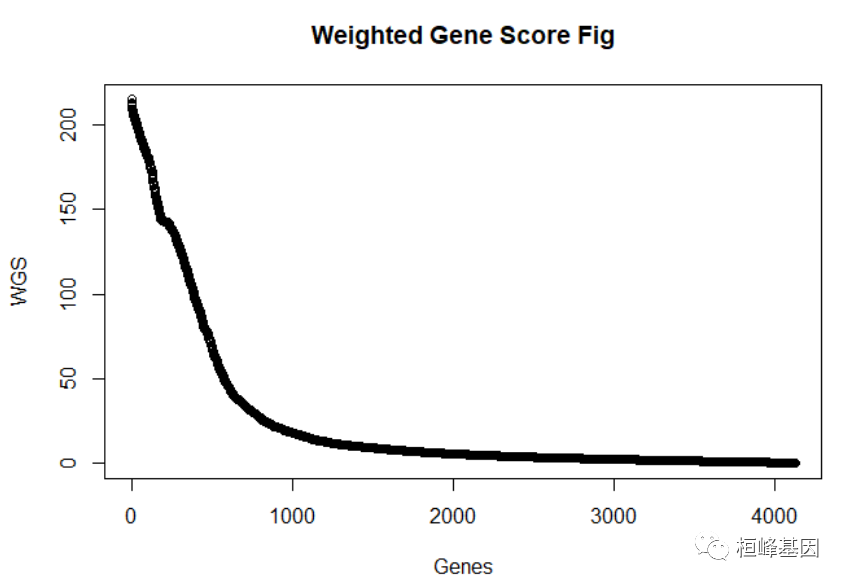
head(wgs)
## WGS
## ENSG00000142959 12.922278
## ENSG00000163815 5.692585
## ENSG00000107611 142.975596
## ENSG00000162461 54.616474
## ENSG00000163959 15.600301
## ENSG00000144410 142.944902
2. intramodularconnectivity{WGCNA}
关键模块和hub基因筛选,在流程中并不可知模块划分好后如何找到key module
-
由WGCNA得到的module都进行GO或KEGG,甚至TF,miRNA等的富集分析,找出所研究性状相关通路相关性最强的module,深入进行研究;
-
看自己感兴趣的gene位于哪个模块,进而去查看;
-
模块与性状的相关性,这个流程中说了,相关性越强,越值得研究。
我们可以通过如下方式获得:
-
High intramodular k within the module(KIM)
-
High module membership (KMM,表达值与ME高相关)
这个用的相对多,因为容易计算,有p值,可跨module比较。这个只能作为继续研究的指导,因为很多gene有非常相似的kME,都可以认为hub gene,还是需要借助外部信息,经验等。ranking应该作为一个粗略建议,所以相似的ranking应看做等价。Top ranked gene应该使用已有的先验知识进行过滤,假如对某个gene感兴趣,不要在乎它是第1还是第3。
- mdoule membership(MM)
最后模块的选择方式,如下:
MM = as.data.frame(cor(datExp, MEs, use = "p"))
对于模块内部的选择方式,如下:
KIM = intramodularconnectivity(adjacency, moduleColors, scaleByMax = TRUE)
文章解读
我们发现好多文章直接就是识别 Hub基因为最终目标而直接发表文章,说明这部分的分析同样也是 SCI 文章中一个热点模块,经过咱们的介绍之后就可以自己动手做起来了。Hub 基因其实就是转录组做完差异分析之后,获得的基因有非常多,我们可以进一步筛选,可以通过WGCNA的方式找到显著模块,进而在显著模块做找到Hub基因,这样就可以缩小范围,进行后续的分析,少量的基因又可以方便后续的实验室验证,增加可行度,达到文章的完整性。

我们可以看下这篇文章的分析流程,都包括哪些数据以及做了哪些分析?
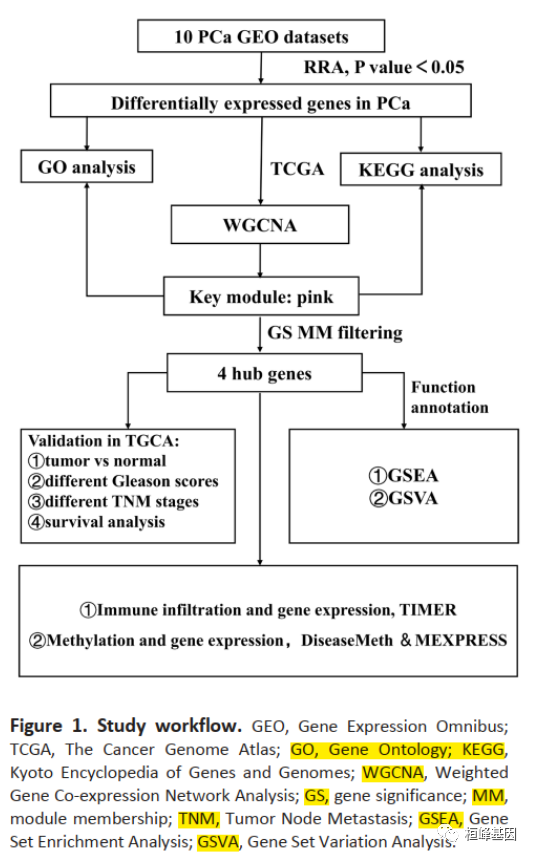
首先是数据库选择:
a. 10个GEO数据集
b. 1个TCGA数据集
分析方法都是常规基于RNA的分析套路:
a. RRA差异分析
g. GSVA 基因集变异分析
j. 结合甲基化数据联合分析hub基因
最后结合临床信息得出一定的结论就完成这篇文章了,在这篇文章里的分析方法基本都涵盖了,小伙伴们可以对应着进入公众号的文章,对照着做了!
References:
-
Yu H, Kim PM, Sprecher E, Trifonov V, Gerstein M (2007) The Importance of Bottlenecks in Protein Networks: Correlation with Gene Essentiality and Expression Dynamics. PLoS Comput Biol 3(4): e59.
-
Song ZY, Chao F, Zhuo Z, Ma Z, Li W, Chen G. Identification of hub genes in prostate cancer using robust rank aggregation and weighted gene co-expression network analysis. Aging (Albany NY). 2019;11(13):4736-4756.
-
Wang J, Chen L, Wang Y, Zhang J, Liang Y, Xu D (2013) A Computational systems biology study for understanding salt tolerance mechanism in Rice. PLoS one 8(6): e64929.

















 1746
1746

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









